2021.06.02.4
Files > Volume 6 > Vol 6 No 4 2021 > Vol 6 No 2 2021
INVESTIGATION / RESEARCH
Soluble production of a full-length human papillomavirus type 16 L1 protein by Escherichia coli

Yunier Serrano1, Susana Miraidys Brito, Elsa Pimienta1, Alina Falero1, Karen Marrero1
Available from: http://dx.doi.org/10.21931/RB/2021.06.02.4
ABSTRACT
Persistent infection with human papillomavirus type 16 (HPV16) causes the development of cervical cancer. Escherichia coli is a cost-effective host successfully used to develop a second-generation vaccine against HPV, based on the purification of soluble truncated L1 protein variants. Previous attempts to produce soluble full-length HPV16-L1 protein by E. coli have failed. This study was aimed at cloning a Cuban HPV16-L1 gene in E. coli and assessing its expression as a soluble full-length L1 protein by manipulating culture conditions. The L1 gene was amplified from a Cuban patient’s cervical sample and cloned into pET28a and pBAD/Myc-HisA vectors. Production and solubility of L1 protein were evaluated in E. coli TOP10 harboring pBADHPV16-L1 plasmid and E. coli BL21-(DE3), Rosetta-(DE3)/pLysS, and SHuffle® T7 Express lysY strains harboring pETHPV16-L1 plasmid, grown under arabinose (0.2%)- or isopropyl β-D-1-thiogalactopyranoside (IPTG, 100 µM)-induction or Super Broth-based auto-induction for 24 and 48 h. The recombinant plasmids pETHPV16-L1 and pBADHPV16-L1 were constructed. The HPV16-L1 protein was produced insoluble to high levels in conventionally IPTG-induced E. coli-pETHPV16-L1 cells. However, under auto-induction, soluble full-length HPV16-L1 protein was successfully produced at similar levels by E. coli BL21 (DE3), Rosetta (DE3) pLysS and SHuffle® T7 Express lysY cells, reaching up to 7.2 ± 0.5% and 14.3 ± 1.6% of the total proteins in the soluble fraction after growing for 24 and 48 h, respectively. It is concluded that the auto-induction procedure at 18 °C with 30 µM IPTG and 100 rev/min promotes soluble full-length HPV16-L1 protein production by E. coli.
Keywords: Human papillomavirus type 16, Escherichia coli, L1 protein
INTRODUCTION
Human papillomavirus (HPV) are non-enveloped double-stranded DNA viruses 1, of which more than 200 types have been described as pathogens in humans 2. HPVs are classified into high-risk (H) and low risk (LR) according to their oncogenic potential 2. The persistent infection of approximately 15 high-risk HPV types causes almost all cervical cancer cases (CC) and immediate precursor lesions 3. Of these genotypes, HPV16 and HPV18 account for approximately 70% of global CC cases 4. As a whole, the CC is the third most common carcinoma among women 5. In particular, HPV16 is the most prevalent high-risk genotype globally 6,7, and studies have shown that it is related to a large proportion of other HPV-associated cancers worldwide 7.
The papillomaviruses consist of L1 and L2 structural proteins, being L1 protein the preferred target for HPV vaccine development due to its self-assembling into virus-like particles (VLPs) mimic the structure of native virions 4,8. The L1-VLPs retain the vast majority of the natural virus’s neutralizing epitopes and can induce high titers of neutralizing antibodies 4. There are currently three HPV-VLP-based prophylactic vaccines available: Cervarix, Gardasil-4, and Gardasil-9 3. Clinical trials have shown that these vaccines induced neutralizing and protective antibodies to prevent HPV-associated infections 7. However, the vaccines’ high production and delivery costs are significant barriers to global implementation, mainly in low-incoming countries where cervical cancer results in higher mortality 9. In Cuba, the CC constitutes the fourth cause of cancer deaths in women 10, being the HPV16 the most frequently found genotype 5. The molecular epidemiologic results support the national implementation of vaccination against HPV in our country 5,11, unfeasible at present due to the high costs of current vaccines. Thus, there is a pressing need for more cost-effective vaccines 1.
The bacterium Escherichia coli constitutes the most attractive host for recombinant protein production due to their low cost, high productivity, and rapid production rates 12, 13. Using this host, a VLP-based vaccine was recently licensed in China 14. L1 proteins in this vaccine (including HPV16-L1 protein) were produced in truncated form and purified from E. coli soluble fraction 3,15. However, soluble full-length HPV16-L1 protein production in E. coli has not yet been achieved since L1 protein is mainly detected in the cells induced by the cells’ insoluble fractions IPTG 16. The auto-induction culture is a strategy that allows accumulating soluble recombinant protein in E. coli cytoplasm 17-19, as well as the employ of low temperature and low inductor concentration 19-21. Therefore, it would be attractive to produce high amounts of soluble full-length HPV16-L1 protein, considering that L1 protein purification from soluble fraction may be a cost-effective alternative since refolding of purified protein is avoided 3. Thus, the present study aimed to assess soluble full-length L1 protein production from a wild type HPV16-L1 gene in E. coli by manipulating culture conditions.
MATERIALS AND METHODS
Reagents, E. coli Strains, and Plasmids
Restriction enzymes, T4 DNA ligase and Pfu DNA polymerase, PCR reagents, 1kb DNA Step Ladder, Broad Range Protein Molecular Weight Markers, Wizard® Minipreps DNA purification system, and Wizard® Gel and PCR Clean-Up System were purchased from Promega (USA). Agar, tryptone, yeast extract, and sodium chloride were from OXOID (England). Kanamycin, ampicillin, and chloramphenicol antibiotics were from AppliChem (Germany). IPTG, the auto-induction medium components, and all chemicals used in SDS-PAGE and Western Blot were from Merck (Germany). The E. coli strains and plasmids used in this study are listed in Table 1.
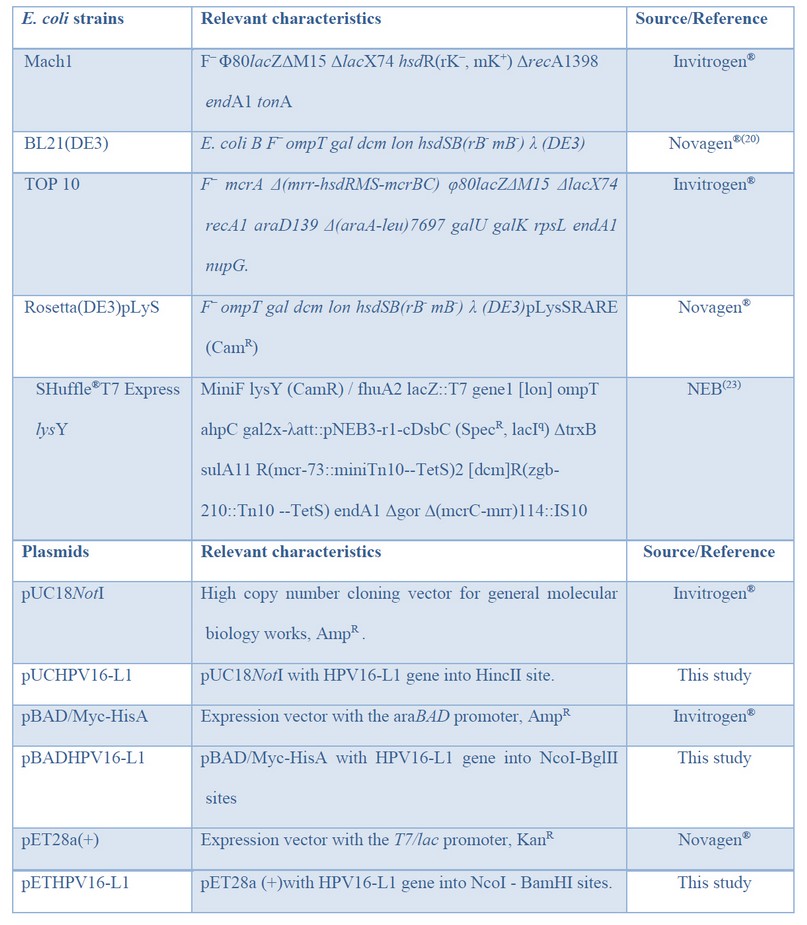
Table 1: Bacteria and plasmids used in this study.
All DNA manipulations, including restriction digestions, ligations, and agarose gel electrophoresis, were performed according to standard procedures 24. Preparation of E. coli competent cells and plasmid transformation protocols were adapted from Li et al. 25.
Amplification of HPV16-L1 gene and cloning into pUC18NotI plasmid
The HPV16-L1 gene (1527 bp) was amplified by Polymerase Chain Reaction (PCR) using total DNA isolated from an HPV16-diagnosed biopsy sample from a Cuban patient 26 and the primer pairs 5’- CCATGGGTCTTTGGCTGCCTAGTG -3’ and 5’- AGATCTCTTACAGCTTACGTTTTTTGCGTT -3’. The amplified gene was flanked by NcoI and BglII sites (underlined) at the 5’ and 3’ ends, respectively, and it was cloned into HincII-digested pUC18NotI vector generating the pUCHPV16-L1 plasmid in E. coli Mach1. Restriction analysis and nucleotide sequencing (Macrogen, Republic of Korea) were used to confirm the HPV16-L1 gene presence in recombinant clones.
Construction of E. coli HPV16-L1 expression plasmids
The wild type HPV16-L1 gene (1518 bp) encoding full-length L1protein from pUCHPV16-L1 was sub-cloned into pET28a (+) and pBAD/Myc-HisA expression vectors as a NcoI-BglII fragment, obtaining the pETHPV16-L1 and pBADHPV16-L1 recombinant plasmids, respectively. These plasmids were characterized by restriction, then pBADHPV16-L1 was introduced into E. coli TOP10, while the pETHPV16-L1 plasmid was transformed into E. coli BL21(DE3), Rosetta (DE3)pLysS, and SHuffle® T7 Express lysY. pBAD/Myc-HisA and pET28a(+) vectors were introduced into E. coli TOP10 and E. coli BL21(DE3), respectively, as expression controls.
SDS-PAGE and Western Blot analysis
For determination of molecular weight, identity testing, and solubility of HPV16-L1 protein, 10% (w/v) sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10 % SDS-PAGE) under reduced conditions 27 was used, followed by staining with Coomassie brilliant blue R250. For Western Blot, all samples were diluted 1:20, except the E. coli TOP10 lysates and electroblotted into a nitrocellulose membrane (Sigma, USA). The HPV16-L1 protein was detected with the MAB885 (CamVir-1) antibody (1:50 000, Merck, Germany). Blots were then treated with goat anti-mouse IgG peroxidase (HP) conjugated (1:64 000, Cuba) and revealed with 3, 3’- diaminobenzidine tetrahydrochloride substrate (Sigma, Germany). The intensity of protein bands from the SDS-PAGE gel was calculated using ImageJ software 19.
Evaluation of HPV16-L1 protein production by E. coli BL21 (DE3) and TOP 10 in test tubes
E. coli TOP10 cells harboring pBAD/Myc-HisA or pBADHPV16-L1 and E. coli BL21(DE3) cells harboring pET28a (+) or pETHPV16-L1 plasmids were inoculated into 5 mL Super Broth medium (SB) (32 g/L of tryptone, 24 g/L of yeast extract, 5 g/L of NaCl, pH 7.2) containing appropriate antibiotics and incubated at 37°C and 200 rev/min for 16 h. These overnight cultures were diluted 100 times in test tubes containing 5 ml of fresh SB medium. The cells were incubated at 37°C until they reached an optical density at 600 nm (OD600 nm) of 0.4–0.6. Then, the expression of L1 gene was induced by the addition of 0.1 mM IPTG or 0.2% (w/v) of L(+)-arabinose. After incubation for an additional 3 h at 37 °C, cells were harvested by centrifugation and suspended in 1x PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) at an OD600nm = 6.5. The 80 μL of cell suspension was immediately mixed with 20μl of SDS-sample buffer (0.25 M Tris-HCl, pH 6.8, 2% SDS, 5% 2-mercaptoethanol, and 5% glycerol) and boiled at 95 °C for 10 min.
Evaluation of HPV16-L1 protein production by E. coli under conventional induction with IPTG in shake flasks
For L1 protein production by E. coli BL21(DE3), Rosetta, and SHuffle containing pETHPV16-L1 plasmid, a single colony from each strain was grown overnight in 5 ml of SB medium-plus kanamycin (50 µg/ml) at 30 °C and 200 rev/min. Overnight cultures were diluted 100 times in fresh SB medium and incubated at 27°C and 200 rev/min until they reached an OD600=0.4-0.6. E. coli cells were then induced with 0.1 mM IPTG for 4 h under the same conditions. E. coli BL21 (DE3) harboring pET28a(+) vector was included as the negative control. Sample preparation was carried out as before for HPV16-L1 protein production in test tubes.
Evaluation of HPV16-L1 protein production by E. coli under auto-induction in shake flasks
Production of L1 protein by E. coli BL21(DE3), Rosetta, and Shuffle strains carrying the pETHPV16-L1 plasmid and E. coli BL21 (DE3) strain harboring pET28a(+) vector were also assayed in an SB-based auto-induction medium 28, using 30 μM IPTG instead of lactose as auto-inducer. Thus, a single colony of each strain was pre-cultured overnight into test tubes containing 5 ml of SB medium with kanamycin (50 µg/ml) at 30 °C and 200 rev/min for 16 h. Then, 2 ml of the grown-culture were transferred into a 2 L- flask containing 200 ml of auto-induction medium supplemented with the same antibiotic. The cultures were incubated at 18 °C and 100 rev/min for 24 or 48h. Sample preparation was carried out as before for HPV16-L1 protein production in test tubes.
Evaluation of HPV16-L1 protein solubility in E. coli under different culture conditions
For assessing HPV16-L1 protein solubility from conventionally-induced cells or auto-induced cells; E. coli BL21(DE3), Rosetta, and SHuffle harboring pETHPV16-L1 plasmid were harvested by centrifugation at 10 000 rev/min, 10 min, 4°C (5810R, Eppendorf AG, Germany), washed twice with 50 mL of 1 x PBS and suspended in 40 ml of 1 x PBS. Then, bacterial cells were lysed using an Emulsiflex C3 cell disrupter (Avastin, Germany), and 80 µL of each cell lysate was saved as the whole cell’s protein (WCP). The samples’ remainder was centrifuged (10,000 rev/min, 10 min) to separate soluble fractions (SF).
Bioinformatic tools
Gene Runner 3.05 Version was used for PCR primer design, and vector NTI Suite 7 was employed for plasmids restriction analyses.
Statistical analysis
All experiments were performed at least three times. Determination of statistical differences was performed with Prism 6 (Graphpad Software, Inc., San Diego, CA, USA) using one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test. Error bars presented as the means ± standard deviation (S.D.).
RESULTS
Cloning of HPV16-L1 gene and construction of pETHPV16-L1 and pBADHPV16-L1 expression plasmids
PCR amplification using total DNA isolated from a Cuban patient’s cervical sample showed a fragment of ~1.5 kb (Figure 1A), close to the expected value (1527 bp). The amplified L1 gene codes for a 57 kDa full-length HPV16-L1 protein. The amplified fragment was then cloned into the pUC18NotI vector obtaining the pUCHPV16-L1 recombinant plasmid, which was subsequently sequenced. Analysis of cloned-L1 gene nucleotide sequence by Blastn showed it was 99% identical to the L1 gene from the HPV16 reference strain (accession number: K027118). Nucleotide sequencing identified guanine (G) at 604 positions instead of cytosine (C) in the L1 gene of the HPV16 reference strain, thus resulting in a change of CAT codon histidine-202 in the L1 gene of HPV16 reference strain by GAT codon for aspartate-202 in the cloned L1 gene. The second GGT codon, coding for glycine-2, was identified in the cloned L1 gene resulting from the insertion of the NcoI site required for subsequent sub-cloning into pET28a (+) and pBADMyc-HisA expression vectors.
After HPV16-L1 gene sequencing, it was subcloned into pBAD/Myc-HisA and pET28a(+) vectors obtaining the pBADHPV16-L1 and pETHPV16-L1 expression plasmids. L1 presence in recombinant plasmids was verified by double restriction with NcoI and HindIII enzymes and by single restriction with BamHI enzyme. Said digestions yielded DNA fragments with sizes close to the theoretical ones and showed that L1 gene was correctly inserted under araBAD (Figure 1 B, lanes 2-3) and T7/lac promoter control (Figure 1 C, lanes 2-3).
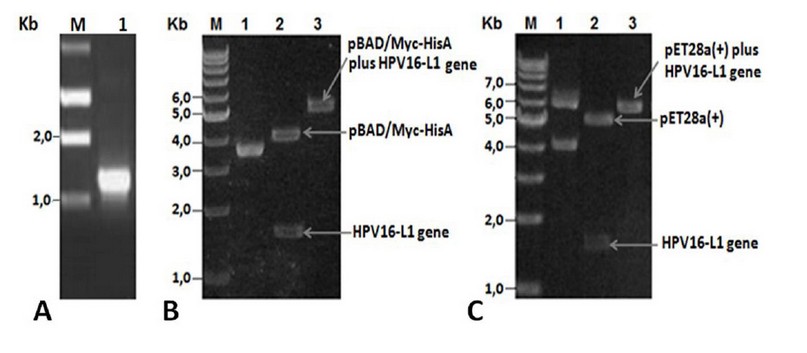
Figure 1. A) PCR product analyses. Lane M: 1Kb DNA Step ladder (Promega), 1: Amplified wild-type HPV16-L1 gene. B) pBADHPV16-L1 plasmid restriction analyses. Lane 1: undigested pBADHPV16-L1 plasmid, lane 2: NcoI-HindIII digested pBADHPV16-L1 plasmid, lane 3: BamHI digested pBADHPV16-L1 plasmid. C) pETHPV16-L1 plasmid restriction analyses. Lane 1: undigested pETHPV16-L1 plasmid, lane 2: NcoI-HindIII digested pETHPV16-L1 plasmid, lane 3: BamHI digested pETHPV16-L1 plasmid.
HPV16-L1 protein production by E. coli BL21 (DE3) and TOP 10 strains
The IPTG-induced E. coli BL21 (DE3)/pETHPV16-L1 cells produced HPV16-L1 protein as an intense band of about 57 kDa (Figure 2A, lane 2), which was recognized, along with other smaller species, by the HPV16-L1 specific monoclonal antibody CamVir (Figure 2B, lane 2). In contrast, arabinose-induced E. coli TOP 10 cells produced low levels of HPV16-L1 protein (Figure 2A, lane 4), only detected by Western blotting with CamVir antibody (Figure 2B, lane 4).
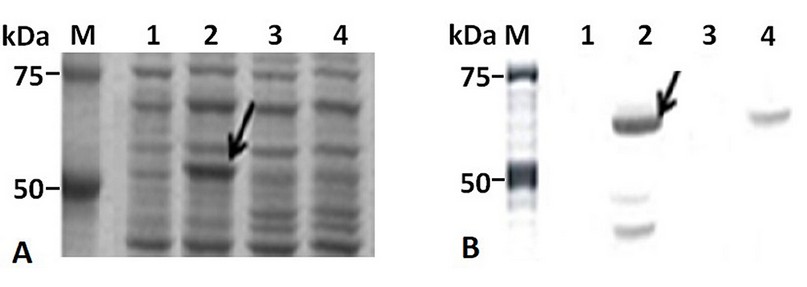
Figure 2. HPV16-L1 protein production by E. coli from pBAD/Myc-HisA or pET28a-derived plasmids. A): 10% (w/v) SDS-PAGE and B): Western Blot with CamVir-1 antibody of IPTG- or arabinose-induced-cells. Lane M: Broad Range Molecular Weight Marker (Promega), lane 1 and 2: E. coli BL21(DE3) harboring pET28a(+) and pETHPV16-L1 plasmids, respectively. Lane 3 and 4: E. coli TOP10 harboring pBAD/Myc-HisA and pBADHPV16-L1 plasmids, respectively.
HPV16-L1 protein production by IPTG-induced or auto-induced-grown E. coli BL21 (DE3), Rosetta, and SHuffle strains in shake flasks
HPV16-L1 protein production was evaluated in BL21 (DE3), Rosetta, and Shuffle strain transformed with pETHPV16-L1 plasmid under IPTG auto-induction. E. coli BL21 (DE3), Rosetta, and SHuffle cells produced HPV16-L1 protein as an intense band of about 57 kDa under IPTG-induction (Figure 3A, lanes 2, 3, and 4) as well as under auto-induction for 24 h (Figure 3D; lanes 2, 3 and 4) and 48 h (Figure 3G; lanes 2, 3 and 4). The overproduced protein under all tested culture conditions, as well as smaller species, reacted with the HPV16-L1 specific CamVir antibody (Figure 3B, E and H; lanes 2, 3 and 4). The BL21(DE3) lysates carrying the pET28a (+) vector showed a weak band (Figure 3A, D and G: lane 2) that CamVir did not recognize (Figure 3B, E and H: lane 2).
Conventionally-IPTG-induced cells produced similar amounts of L1 protein (Figure 3C). However, under auto-induction conditions for 24 h, BL21(DE3) produced the highest amount of L1 protein concerning Rosetta (p<0.0001) and SHuffle (Figure 3F; p<0.01). For 48 h, BL21(DE3) also produced the highest amount of L1 protein compared to Rosetta (p<0.0001) and SHuffle (Figure 3I; p<0.05). The L1 protein level was higher in SHuffle regarding Rosetta at 48h (Figure 3I; p<0.01).
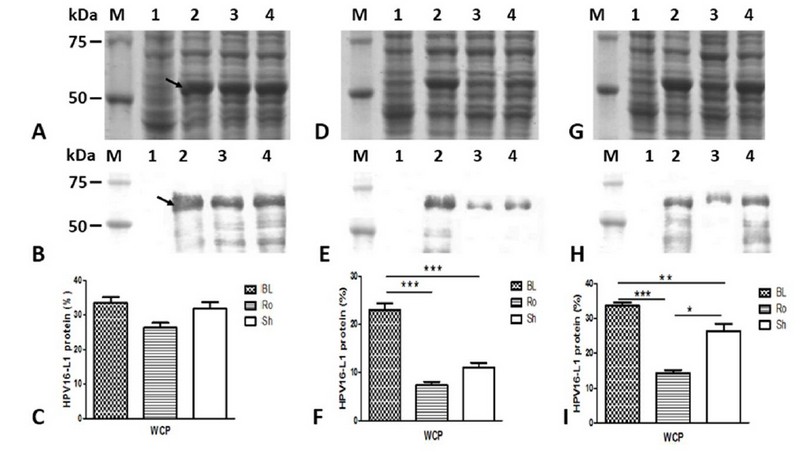
Figure 3: Production of HPV16-L1 protein by E. coli BL21(DE3), Rosetta, and SHuffle strains harboring pETHPV16-L1 plasmid grown under different conditions. A, D, and G: 10 % ( w/v) SDS-PAGE of 4h-IPTG-induced, 24h-auto-induced, and 48h-auto-induced cells, respectively. B, E, and H: Western Blot with CamVir -1 antibody (1: 50 000) of 4h-IPTG-induced, 24h-auto-induced, and 48h-auto-induced cells, respectively. Lane M: Broad Range Protein Molecular Weight Markers (Promega), lane 1: BL21(DE3)/pET28a(+) vector, Lane 2: BL21(DE3)/pETHPV16-L1, Lane 3: Rosetta /pETHPV16-L1, Lane 4: SHuffle /pETHPV16-L1. Arrows indicate L1 protein. C, F and I: Densitometric analyses of SDS-PAGE scanned gels. Statistical analyses were performed by one-way ANOVA followed by Tukey’s multiple comparisons test. Significant differences were considered at P< 0.05, P < 0.01 and P<0.0001. The data are presented as means ± SD (N = 3). BL: BL21(DE3), Ro: Rosetta, and Sh: SHuffle.
Solubility of HPV16-L1 protein produced by IPTG-induced- or auto-induced-grown E. coli BL21 (DE3), Rosetta, and SHuffle strains in shake flasks
The HPV16-L1 protein’s accumulation was undetected in the soluble fractions of the conventionally-IPTG-induced cells from the three E. coli evaluated strains (Figure 4A and B: lanes 2, 4, and 6). By contrast, in auto-induced cells, the soluble L1 protein was detected (Figure. 4 D and G: lanes 2, 4, and 6) at similar levels by the three E. coli strains (Figure 4F and I). The percentage of HPV16-L1 protein concerning the total proteins in auto-induced cells during 24 and 48 h was 7.2 ± 0.5% and 14.3 ± 1.6%, respectively. Moreover, part of L1 protein was produced insoluble in BL21 auto-induced for 24 h (Figure 4 D and E: lanes 1 and 2), as well as in BL21(DE3) and SHuffle auto-induced for 48 h (Figure 4G and H: lanes 1,2,5 and 6). However, in Rosetta grown for 24 h or 48 h, practically all the produced L1 protein accumulated in the soluble fraction (Figure 4 D and G: lanes 3 and 4).
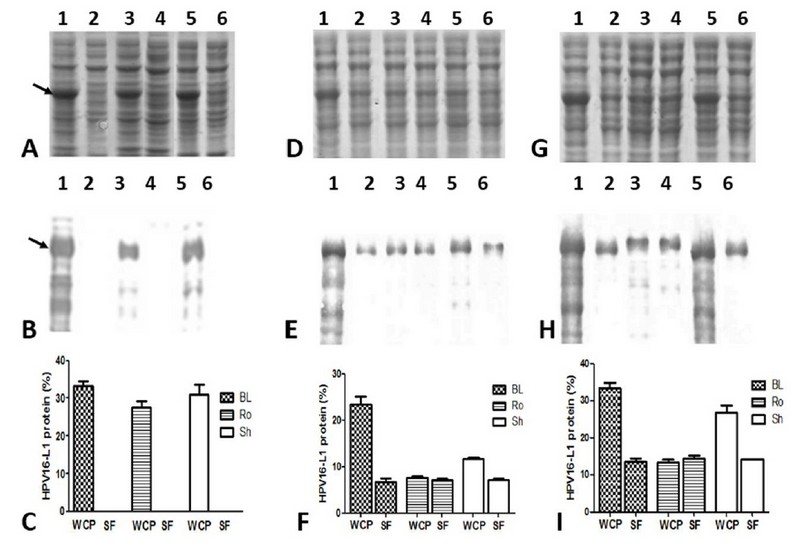
Figure 4: Solubility of HPV16-L1 protein in E. coli BL21(DE3), Rosetta, and SHuffle strains harboring pETHPV16-L1 plasmid grown under different conditions. A, D, and G: 10 % SDS-PAGE of 4h-IPTG-induced, 24h-auto-induced, and 48h-auto-induced cells, respectively. B, E, and H: Western Blot with CamVir-1 antibody (1: 50 000) of 4h-IPTG-induced, 24h-auto-induced, and 48h-auto-induced cells, respectively. Lane 1: BL21(DE3) whole cell lysate, 2: BL21(DE3) soluble fraction, 3: Rosetta whole cell lysate, 4: Rosetta soluble fraction, 5: SHuffle whole cell lysate, 6: SHuffle soluble fraction. Arrows indicate L1 protein. * Whole Cell Protein (WCP), Soluble Fraction (SF). C, F and I: Densitometric analyses of SDS-PAGE scanned gels. Soluble HPV16-L1 protein percent among different strains was not different, by one-way ANOVA followed by the Tukey’s multi comparisons test. Data presented as means ± SD (N = 3). BL: BL21(DE3), Ro: Rosetta, and Sh: SHuffle.
DISCUSSION
The effectiveness of commercially available anti-HPV VLPs-based vaccines has promoted a considerable interest in developing vaccine candidates from cheaper hosts 8,29. VLP-based vaccines derived from E. coli are more cost-effective than those prepared from insect cells or yeasts at an industrial scale. (1) A VLP-based vaccine assembled from soluble N-terminal truncated variants of L1 protein in E. coli was developed 3, 15 and recently licensed in China 14, showing this host’s potential for producing L1 proteins. In the present study, a wild-type full-length HPV16-L1 gene was amplified by PCR from total DNA extracted from a biopsy sample of an HPV16-diagnosed patient with cervical cancer amplified gene was cloned and verified by sequencing. The cloned HPV16-L1 gene matches the European variant (Accession K02718) with the highest sequence homology level (99 %). There was only one nucleotide difference between the K02718 (C) and the cloned sequence (G) at position 604, changing the codon CAT for histidine-202 in the reference sequence to GAT codon encoding aspartate-202 in the amplified product, which otherwise is required for VLPs assembly in recombinant systems 30.
Successful expression and solubility of recombinant proteins depend on, among the main factors: the choice of the expression vector, expression host, medium composition, and induction conditions 13, 20. Thus, it was assessed the production level and solubility of full-length HPV16-L1 protein in E. coli TOP 10 transformed with the pBADHPV16-L1 plasmid and E. coli BL21(DE3), Rosetta and SHuffle transformed with pETHPV16-L1 plasmid by conventional induction with L(+)-Arabinose or IPTG, as well as SB-based auto-induction. L1 protein was efficiently produced in BL21(DE3) induced with IPTG employing the pET vector series previously used to produce the HPV16-L1 protein in E. coli host by other researchers 31. The smaller species recognized by CamVir-1 antibody in BL21(DE3) cell lysates agrees with previous reports on HPV16-L1 protein fragmentation from wild-type L1 gene expressed in insect cells 30, Saccharomyces cerevisiae 32 and E. coli cells 8,31.
In contrast to BL21 (DE3)/pETHPV16-L1, the E. coli TOP10/pBAD expression system did not efficiently produce L1 protein. The arabinose-regulated promoter has been widely used for protein expression in E. coli 18 to promote high amounts of soluble recombinant proteins 33. However, in our study, L1 protein was barely detected by Western Blot, which could be related to L1 gene expression’s lesser efficiency from the pBAD vector series. Our results showed that the produced protein corresponds to the L1 protein from HPV16 and that the BL21(DE3)/pET expression system is better than TOP10/pBAD system for the production of L1 protein from the wild-type HPV16-L1 gene.
Once we verified the production of the L1 protein by BL21(DE3) /pET expression system, we proceeded to assay its production in E. coli BL21(DE3), Rosetta, and SHuffle carrying the pETHPV16-L1 plasmid under induction with IPTG or SB-based auto-induction. The results obtained showed that the HPV16-L1 protein was produced at similar levels in conventionally-induced cells. In contrast, the L1 protein production levels in auto-induced cells were significantly different. BL21(DE3) produced the highest amount of HPV16-L1 protein, followed by SHuffle and Rosetta. These results are consistent with Zarschler et al., who found a slightly higher production of single-domain antibodies in BL21(DE3) than SHuffle using the pET system after induced with IPTG 34. The behavior in the production levels of HPV16-L1 protein in SHuffle and Rosetta concerning BL21 (DE3) may be related to constitutive lysozyme production and accumulation within cells. The lysozyme is a natural inhibitor of T7 RNA polymerase (T7 RNAP) 20, which is supplied by pLysS plasmid (10-12 copies per cell) 23 in Rosetta 35 or by miniF plasmid (1-2 copies per cell) 36 in SHuffle 37. It is possible that, during long time-culture (like auto-induction) of lysozyme expressing cells, the lysozyme produced reaches high levels and decreases the T7 RNAP availability, affecting the L1 protein yield in those cells.
The solubility of L1 protein produced by conventionally-induced cells or auto- induced cells was also analyzed. The soluble HPV-L1 protein production might be advantageous for developing a successful and efficient E. coli-based HPV-LI VLPs vaccine 3, due to the soluble protein may preserve the protective conformational epitopes 3,15. In this regard, soluble full-length L1 protein has not been detected by SDS-PAGE from IPTG-induced E. coli cell 31. Likewise, in our study, the L1 protein was found insoluble in conventionally-induced cells, which differs from the soluble production of HPV11-L1 protein from a wild-type L1 gene using the pET vector series 38. By contrast, HPV16-L1 protein solubility was enhanced in auto-induced cells, which is in line with previous reports about using auto-induction as a strategy to promote soluble recombinant protein production in E. coli 17-19. This strategy successfully increased the solubility of recombinant proteins produced insoluble under IPTG-induction in E. coli, such as human tissue plasminogen (tPA)17,28 and streptococcal cysteine protease 39. Long et al. reported soluble production of full-length tPA in E. coli, showing that auto-induction is a reliable strategy to reach at least 50% of tPA expression in the soluble fraction E. coli 17. Three years later, Fathi-Roudsari et al. optimized the expression conditions for the tPA protein (truncated variant) E. coli by decreasing temperature and using an SB-based auto-induction medium, resulting in at least three times higher levels of active reteplase production in oxidative cytoplasm of Rosetta-gami 19. Like Fathi-Roudsari et al., we used an SB-based auto-induction medium and 18 ⁰C grown temperature instead of using lactose; a low IPTG concentration (30 µM) was used, which previously has been used efficiently in auto-induction 40. Our results are also consistent with the positive effect of decreasing inducer’s concentration and induction temperature on the recombinant protein solubility in the E. coli cytoplasm 19-21. Thus, a decrease in temperature during induction causes a reduction in the recombinant protein synthesis rate and a reduction in hydrophobic interactions and consequently prevents the formation of inclusion bodies 20. Fathi-Roudsari et al. found that decreasing the temperature to 25 and 18 °C increased active reteplase level by 20 and 60%, respectively 19.
On the other hand, the amount of L1 protein accumulated in BL21(DE3), Rosetta, and SHuffle’s soluble fraction was similar, suggesting that soluble L1 protein production depends more on culture conditions than the genetic background of each strain (Table 1) 20,23,35. Gu et al. produced soluble truncated HPV16-L1 protein with a purity of about 10% under conventional induction with IPTG 41, a level similar to those achieved in the current study (7.2 and 14 % 24 and 48 h of autoinduction, respectively).
Our results demonstrated that a slow expression procedure that combines growth at a low temperature, a low IPTG concentration, and a low agitation speed under auto-induction conditions promotes soluble HPV16-L1 protein accumulation. These constitute the first report describing the successful production of soluble full-length HPV16-L1 protein from a wild-type L1 gene in E. coli using an auto-induction strategy. This could be valuable for using prokaryotic auto-induction expression systems to produce HPV16-L1 antigen for vaccine development, omitting the in vitro refolding step for its purification. Purification of the soluble full-length HPV16-L1 protein for structural and immunogenicity analyses is currently underway.
In short, a Cuban wild type full-length HPV16-L1 gene was successfully cloned and expressed in E. coli BL21(DE3), Rosetta (DE3) pLysS, and SHuffle® T7 Express lysY employing the pET system. E. coli culture under auto-induction at 18°C, with 30µM IPTG and a low agitation (100 rev/min) promotes soluble production of full-length HPV16-L1 protein. development of this bacterial system could be used for purifying HPV16-L1 protein from soluble fraction and evaluating its capacity to form VLPs, which could be used as vaccine candidate antigen.
CONCLUSION
The auto-induction procedure at 18 °C with 30 µM IPTG and 100 rev/min promotes soluble full-length HPV16-L1 protein production by E. coli.
Acknowledgment
We thank Dr. Juan Carlos Piña for providing us with the genomic DNA for HPV16L1 gene amplification.
Conflict of Interest
The authors declare that they have no conflict of interest.
REFERENCES
1. Huang X, Wang X, Zhang J, Xia N, Zhao Q. Escherichia coli - derived virus-like particles in vaccine Development. npj Vaccines. 2017; 2(3):1-9.
2. Harlé A, Guillet J, Thomas J, Sastre-Garau X, Rouyer M, Ramacci C, et al. Evaluation and validation of HPV real-time PCR assay for the detection of HPV DNA in oral cytobrush and FFPE samples. Scientific Reports. 2018; 8:11313.
3. Wei M, Wang D, Li Z, Song S, Kong X, Mo X, et al. N-terminal truncations on L1 proteins of human papillomaviruses promote their soluble expression in Escherichia coli and self-assembly in vitro. Emerg Microbes Infect. 2018; 7(1):160.
4. Zhang Y, He Y, Li L, Liang S, Yan M, Ren D, et al. development and characterization of an HPV18 detection kit using two novel HPV18 type-specific monoclonal antibodies. Diagnostic Pathology. 2018; 13:55.
5. Soto Y, Torres G, Kourí V, Limia CM, Goicolea A, Capó V, et al. Molecular epidemiology of human papillomavirus infections in cervical samples from cuban women older than 30 years. J Low Genit Tract Dis. 2014; 18(3):210-217.
6. Javanmard D, Namaei M H, Haghighi F, Ziaee M, Behavan M, Mirzaei J, et al. The Frequency and Typing of Human Papilloma Virus Among Women with Normal and Abnormal Cytology in Southern Khorasan, Eastern Iran. Jundishapur J Microbiol. 2017; 10(4): e43213.
7. Maleki Z. Human papilloma virus vaccination: Review article and an update. World J Obstet Gynecol. 2016; 5(1): 1627.
8. Bang HB, Lee YH, Lee YJ, Jeong KJ. High-Level Production of Human Papillomavirus (HPV) Type 16 L1 in Escherichia coli. J Microbiol Biotechnol. 2016; 26(2):356-63.
9. Bruni L, Diaz M, Barrionuevo-Rosas L, Herrero R, Bray F, Bosch FX, et al. Global estimates of human papillomavirus vaccination coverage by region and income level: a pooled analysis. Lancet Glob Health. 2016; 4(7):e453–63.
10. Bruni L, Albero G, Serrano B, Mena M, Gómez D, Muñoz J, Bosch FX, de Sanjosé S. ICO/IARCInformation Centre on HPV and Cancer (HPV Information Centre). Human Papillomavirus and RelatedDiseases in Cuba. Summary Report 17 June 2019. Available from: https://hpvcentre.net/statistics/reports/CUB.pdf
11. Guilarte E, SotoY, Kourí V, Limia CM, Sánchez ML, Rodríguez AE, et al. Circulation of human Papillomavirus and Chlamydia trachomatis in women. MEDICC Rev. 2020;22(1):17–27.
12. Baeshen MN, Al-Hejin AM, Bora RS, Ahmed MM, Ramadan HA, Saini KS, et al. Production of Biopharmaceuticals in E. coli: Current Scenario and Future Perspectives. J Microbiol Biotechnol. 2015; 25 (7): 953-62.
13. Uhoraningoga A, Kinsella GK, Henehan GT, Ryan BJ. The Goldilocks Approach: A Review of Employing Design of Experiments in Prokaryotic Recombinant Protein Production. Bioengineering (Basel). 2018; 5(4):89.
14. Qian C, Liu X, Xu Q, Wang Z, Chen J, Li T, Zheng Q, et al. Recent Progress on the Versatility of Virus-Like Particles. Vaccines. 2020; 8(139):1-14.
15. Gu Y, Wei M, Wang D, Li Z, Xie M, Pan H, et al. Characterization of an Escherichia coli-derived human papillomavirus type 16 and 18 bivalent vaccine.Vaccine. 2017; 35(35 Pt B):4637-4645.
16. Kelsall SR, Kulski JK. Expression of the major capsid protein of human papillomavirus type 16 in Escherichia coli. J Virol Methods. 1995; 53(1):75-90.
17. Long X, Gou Y, Luo M, Zhang S, Zhang H, Bai L, et al. Soluble expression, purification, and characterization of active recombinant human tissue plasminogen activator by auto-induction in E. coli. BMC Biotechnology. 2015;15:13.
18. Jia B, Jeon CO. 2016. High-thoughput recombinant protein expression in Escherichia coli: current status and future perspectives. Open Biol. 2016; 6: 160196.
19. Fathi-Roudsari M, Maghsoudi N, Maghsoudi A, Niazi S, Soleiman M. Auto-induction for high level production of biologically active reteplase in Escherichia coli. Protein Expr Purif. 2018; 151:18-22.
20. Hayat SMG, Farahani N, Golichenari B, Sahebkar A. Recombinant Protein Expression in Escherichia coli (E.coli): What We Need to Know?.Curr Pharm Des. 2018; 24(6):718-725.
21. Yousefi M, Farajnia S, Mokhtarzadeh A, Akbari B, Ahdi Khosroshahi S, Mamipour M, et al. Soluble Expression of Humanized Anti-CD20 Single Chain Antibody in Escherichia coli by Cytoplasmic Chaperones Co-expression. Avicenna Journal of Medical Biotechnology. 2018; 10(3):141-146.
22. Srimeenakshi S, Murthy M, Selvakumar K, et al. An Overview of the Parameters for Recombinant Protein Expression in Escherichia Coli. J Cell Sci Ther. 2015; 6: 221. doi:10.4172/2157-7013.1000221.
23. Rosano GL, Ceccarelli EA. Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol. 2014; 5:172.
24. Zhou Y, Zhang Y, He W, Wang J, Peng F, Huang L, et al. Rapid Regeneration and Reuse of Silica Columns from PCR Purification and Gel Extraction Kits. Sci Rep. 2018; 8(1):12870.
25. Li X, Sui X, Zhang Y, Sun Y, Zhao Y, Zhai Y, et al. An improved calcium chloride method preparation and transformation of competent cells. African Journal of Biotechnology. 2010; 9(50): 8549-8554.
26. Piña JC, Crespo G, Fando RA, Casanova G, Curbelo M, Guerra MM. Identificación molecular de genotipos papilomavirus humanos en pacientes con cáncer de cuello uterino. AMC. 2016; 20( 3 ): 288-298.
27. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970; 227(5259):680-685.
28. Studier FW. Protein production by autoinduction in high density shaking cultures. ProteinExpr. Purif. 2005; 41: 207–234.
29. Guan J, Bywaters SM, Brendle SA, Ashley RE, Makhov AM, Conway JF, et al. Cryoelectron Microscopy Maps of Human Papillomavirus 16 Reveal L2 Densities and Heparin Binding Site. Structure. 2017; 25(2):253-263.
30. Kirnbauer R, Taub J, Greenstone H, Roden R, Dürst M, Gissmann L, et al. 1993. Efficient self-assembly of human papillomavirus type 16 L1 and L1-L2 into virus-like particles. J. Virol.1993; 67:6929-6936.
31. Zhang W, Carmichael J, Ferguson J, Inglis S, Ashafian H, Stanley M. Expression of human papillomavirus type 16 L1 protein in Escherichia coli: denaturation, renaturation, and self-assembly of virus-like particles in vitro.Virology.1998; 243(2):423-31.
32. Patel MC, Patkar KK, Basu A, Mohandas KM, Mukhopadhyaya R. Production of immunogenic human papillomavirus-16 major capsid protein derived virus like particles.Indian J Med Res. 2009; 130(3):213-8.
33. Kaur J, Kumar A, Kaur J. Strategies for optimization of heterologous protein expression in E. coli: Roadblocks and reinforcements. International Journal of Biological Macromolecules 2018; 106 (2018): 803–822.
34. Zarschler K, Witecy S, Kapplusch F, Foerster C, Stephan H. High-yield production of functional soluble single-domain antibodies in the cytoplasm of Escherichia coli. Microb Cell Fact. 2013; 12:97.
35. Novy R., Drott D., Yaeger K., Mierendorf R. (2001). Overcoming the codon bias of E. coli for enhanced protein expression. Innovations 12 1–3.
36. Nozaki S, Niki H, Ogawa T. Replication initiator DnaA of Escherichia coli changes its assembly form on the replication origin during the cell cycle. J Bacteriol. 2009; 191(15):4807-14.
37. Samuelson JC, Davis TB, Raleigh EA, Southworth MW, Inventors; Nyland Biolabs, Inc., Ipswich, assignee: Expression of toxic genes in vivo in a non-natural host. United States patent US 8.492,117 B2 . 2013 Jul 23.
38. Li M, Cripe TP, Estes PA, Lyon MK, Rose RC, Garcea RL. Expression of the human papillomavirus type 11 L1 capsid protein in Escherichia coli: characterization of protein domains involved in DNA binding and capsid assembly. J Virol. 1997; 71(4):2988–2995.
39. Lane MD, Seelig B. Highly efficient recombinant production and purification of streptococcal cysteine protease streptopain with increased enzymatic activity. Protein Expr Purif. 2016; 121:66–72.
40. Faust G, Stand A, & Weuster‐Botz D. (2015). IPTG can replace lactose in auto‐induction media to enhance protein expression in batch‐cultured Escherichia coli. Engineering in Life Sciences, 15(8), 824-829.
41. Gu Y, Li S, Wei M, Xian Y, Luo W, Xia N, Inventors; Xiamen (CN), assignee: Truncated L1 protein of human papillomavirus type 16. US 2010/0255031 Al. 2010 Oct 7.
Received: 28 November 2020
Accepted: 10 January 2021
Yunier Serrano1, Susana Miraidys Brito, Elsa Pimienta1, Alina Falero1, Karen Marrero1
1Unit for Biological Products Research, Division of Research, Development & Innovation, National Center for Scientific Research, Avenue 25 and 158. Cubanacán, Playa, La Habana, Cuba. P.O. Box 6414.
*Corresponding author: Yunier Serrano
National Center for Scientific Research, La Havana, Cuba
e-mail: [email protected];
Tel: 53-7-2086548