2022.07.01.16
Files > Volume 7 > Vol 7 No 1 2022
Rara patología derivada de un anillo del cromosoma 15. Clínica, genómica e interactoma de proteínas de genes asociados al fenotipo
Rare pathology derived from a ring chromosome 15. Clinical, genomic and protein interactome of genes associated with the phenotype

César Paz-y-Miño1-2-3* , Camila Medranda4, Alejandra Loaiza4, Mishell Ponce4, Paola E. Leone2
, Camila Medranda4, Alejandra Loaiza4, Mishell Ponce4, Paola E. Leone2
1 Academia Ecuatoriana de Medicina. Academia Ecuatoriana de Medicina, calles Sodiro y Valparaíso. Pabellón 4. 170136 Quito, Ecuador https://orcid.org/0000-0002-6693-7344
2 Sociedad Ecuatoriana de Genética Humana.
3 Facultad de Ciencias de la Salud Eugenio Espejo. Universidad UTE. Quito, Ecuador
4 Genomics Lab. Amazonas N39-82 y Pereira. Edificio Casa Vivanco, piso 6.
* Correspondence: [email protected]
Available from: http://dx.doi.org/10.21931/RB/2022.07.01.16
RESUMEN
Se presenta el caso de una paciente de sexo femenino de cuatro años y dos meses de edad, quien consultó por un fenotipo especial: Retraso psicomotor, talla corta, microcefalia, orejas grandes y de implantación baja, frente pequeña, arcos superciliares prominentes, comisura labial abierta, hipertelorismo ocular, cuello corto, hipertelorismo mamario y pectus excavatum. El objetivo de este trabajo es analizar una paciente con rasgos fenotípicos inusuales, a través del examen físico, análisis comparativo con otros casos, y estudios genéticos. El estudio citogenético reveló un cariotipo en mosaico, mos 46,XX,r(15)(q26.3)/46,XX con la presencia del anillo en 83%. El estudio de arrays de mapeo genético identificó la pérdida de 3,5 Mb en 15q26.3. Entre los genes perdidos en la región terminal de 15q, se evidenció una interacción entre sus productos proteicos según el análisis por STRING. Este es el segundo caso de anillo del cromosoma 15 informado en el Ecuador, y sería el centésimo primero en el mundo desde 1966. El fenotipo especial de estos individuos se relaciona con la cantidad de material genético perdido. Los genes implicados en la formación del anillo, así como las proteínas que determinarían estos genes y las relaciones en distintas vías celulares, son analizados in sílico con la finalidad de comprender la fisiopatología de este trastorno. Su diagnóstico es mayoritariamente postnatal por lo cual el enfoque clínico difiere individualmente de acuerdo a los síntomas y signos que se presenten.
Palabras clave. 46,XX,r(15)(q26.3), anillo del cromosoma 15, mosaico, array de mapeo genético, interactoma.
ABSTRACT
The case of a 4-year-two-month-old female patient is presented, who consulted for a special phenotype: psychomotor retardation, short stature, microcephaly, large and low-set ears, small forehead, prominent brow ridges, labial commissure open, ocular hypertelorism, short neck, mammary hypertelorism and pectus excavatum. The objective of this study is to analyze a patient with unusual phenotypic traits, through physical examination, comparative analysis with other cases, and genetic studies. The cytogenetic study revealed a mosaic karyotype, mos 46,XX,r(15)(q26.3)/46,XX with the presence of the ring in 83%. The genetic mapping array study identified the loss of 3.5 Mb in 15q26.3. Among the genes lost in the terminal region of 15q, an interaction between their protein products was evidenced according to the STRING analysis. This is the second case of a ring chromosome 15 reported in Ecuador. And it would be the 101st in the world since 1966. The special phenotype of these individuals is related to the amount of genetic material lost. The genes involved in the formation of the ring, as well as the proteins that determine these genes and the relationships in different cellular pathways, are analyzed in silico in order to understand the pathophysiology of this disorder. Its diagnosis is mostly postnatal, so the clinical approach differs individually according to the symptoms and signs that appear.
Keywords. 46,XX,r(15)(q26.3), ring chromosome 15, mosaic, genetic mapping array, interactome.
INTRODUCCIÓN
Los cromosomas en anillo son anomalías cromosómicas estructurales inestables, producidos por roturas terminales en uno o ambos brazos cromosómicos seguidas de un evento de fusión de los extremos rotos 1, o por un reordenamiento inversión-duplicación-deleción 2, los que pueden desencadenar la pérdida o ganancia de material genético. Los cromosomas en anillo también pueden formarse por la fusión de secuencias subteloméricas como por la fusión entre telómeros, lo que no ocasionaría pérdida de material genético 3, 4, como se observa en pacientes con fenotipo normal, pese al anillo.
En general, el fenotipo de los casos con anillo del cromosoma 15, r(15), presenta retraso del crecimiento, bajo peso al nacer, retraso mental variable, microcefalia, hipertelorismo y fascias triangulares 5.
El cromosoma 15 en anillo constituye una alteración estructural rara, siendo su prevalencia menor de uno en un millón, afectando con más frecuencia al sexo femenino en comparación con el masculino. Se han reportado cien casos alrededor del mundo desde 1966 hasta la fecha, uno de ellos en Ecuador en el año 2018 6-8. El presente caso que informamos constituye el segundo de esta patología en el país.
Si bien el diagnóstico prenatal para este tipo de anormalidades es posible mediante pruebas especializadas como amniocentesis, ultrasonido y análisis de vellosidades coriónicas; los resultados obtenidos con estas pruebas sólo pueden ser reconfirmados después del nacimiento y a lo largo de la infancia evaluando la aparición y desarrollo de características clínicas congruentes con esta enfermedad 9. El fenotipo final dependerá del porcentaje de mosaico del anillo que presente el paciente y de la cantidad delecionada de material genético al momento de la formación del anillo.
El objetivo de este trabajo es describir un nuevo caso de una paciente de cuatro años dos meses con rasgos fenotípicos inusuales, que acudió voluntariamente a realizarse una evaluación genética, estudios citogenéticos y moleculares, con la finalidad de entender su patología y analizar su origen y comportamiento genético.
PRESENTACIÓN DEL CASO
La probandus acude por primera vez a consulta genética a los cuatro años dos meses de edad, es de sexo femenino, fue producto de una tercera gestación. La madre de 27 años y padre de 30 años de edad aparentemente sanos, sin anormalidades conocidas y no consanguíneos.
La paciente proviene de la ciudad de Quito ubicada en la región Sierra del Ecuador; de acuerdo a la ancestría de la población ecuatoriana, la paciente es mestiza 10.
La paciente nació por parto vaginal a las 40 semanas de gestación reportando un peso de 1.134 g, perímetro cefálico de 31 cm y APGAR de 8-10. Asiste a consulta genética por presentar problemas en su fenotipo: Retraso psicomotor y del desarrollo, bajo peso al nacer (14.2 Kg <percentil 10), talla corta (105 cm <percentil 3), microcefalia (48 cm <percentil 10), retraso mental, frente pequeña, hipertelorismo ocular, arcos supraciliares prominentes, orejas grandes y de implantación baja, filtrum corto, labios hipoplásicos, comisura labial abierta, nariz pequeña, cuello corto, hipertelorismo mamario y pectus excavatum (Figura 1).
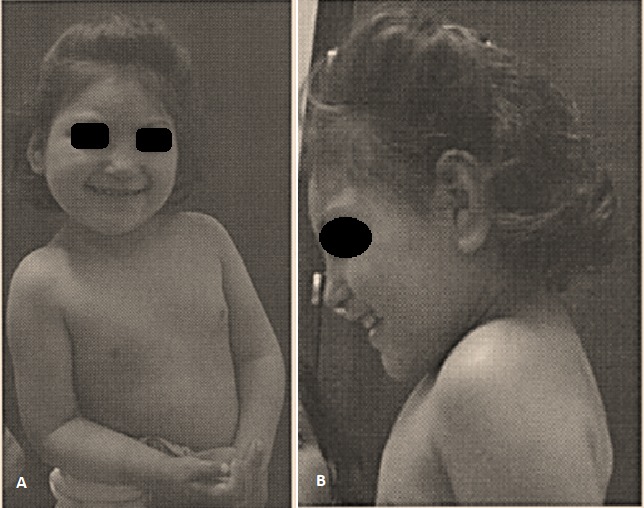
Figura 1. Fotografía de la paciente. A) Vista frontal. B) Vista lateral.
Análisis del fenotipo
Se listó las características físicas de todos los casos con anillo del cromosoma 15 descritos en la literatura 6-8. Se comparó el fenotipo de la paciente con el de la lista de los 100 casos anteriores.
Estudio genético
Tanto a la probandus como a sus padres se les tomó muestras de sangre periférica con heparina y se realizó los estudios citogenéticos utilizando el protocolo estándar del laboratorio; cultivo de 72 horas en medio RPMI-1640, enriquecido con Suero bobino fetal 15%, fitohemaglutinina, antibiótico y antimicótico. Se realizó la cosecha con colchicina y fijador de Carnoy; extensión de portaobjetos y bandeo GTG (Giemsa-Tripsina-Giemsa). Se contabilizaron 100 metafases por individuo.
Para los estudios moleculares se empleó muestras de sangre periférica obtenidas mediante punción venosa y tratadas con ácido etilendiaminotetraacético (EDTA) como anticoagulante. Se utilizaron 200 µl de muestra para la extracción de ADN utilizando el kit comercial Purelink Genomic DNA (Invitrogen®, Life Technologies, CA, USA). La concentración y pureza del material genético se valoró utilizando NanoDrop 2000c Spectrophotometer (Thermo Fisher Scientific Inc., MA, USA) 10.
Se realizó el análisis de los componentes de la ancestría genética, por tipificación de 46 marcadores informativos de ancestría autosómica, basados en inserción y deleción 10. Para describir el estado de las regiones cromosómicas involucradas en el anillo se emplearon arrays de mapeo genético CytoScan 750K (Affymetrix, Santa Clara, CA, USA) según las instrucciones del fabricante (Affymetrix, Santa Clara, CA, USA). Para los genes implicados en la región de pérdida cromosómica se aplicó el análisis bioinformático STRING 11.
RESULTADOS
El análisis de las características físicas descritas en los 100 casos informados en la literatura, reveló 52 rasgos (6-8), 12 de ellos presentes en la actual paciente. La probandus presentó también 6 signos no descritos en otros pacientes (Tabla 1).

Tabla 1. Comparación de los rasgos fenotípicos entre la actual paciente y los 100 casos informados en la literatura con cromosoma 15 en anillo.
El estudio de ancestría de la paciente muestra la siguiente composición étnica: Nativoamericano 62%, Europeo 32% y Afrodescendiente 6%.
El estudio citogenético de la paciente evidenció dos líneas celulares: normal de 46 cromosomas en el 17% de las metafases analizadas y una con la presencia del cromosoma 15 en anillo (Figura 2) en el 83%, con un cariotipo: mos 46,XX,r(15)(q26.3) [83]/46,XX 17. Los cariotipos de los padres fueron normales: 46,XX y 46,XY.

Figura 2. Diferentes tipos de cromosomas 15 en anillo encontrados en la paciente. Cromosoma normal, cromosoma en anillo monocéntrico y cromosoma en anillo dicéntrico. Las fechas señalan los puntos de rotura.
El estudio de arrays de mapeo genético mostró una deleción terminal en 15q26.3 (Figura 3), de 3.459.037 pb con la pérdida de una copia de 29 genes localizados en esta región (desde 98.970.003 pb a 102.429.040 pb) (Tabla 2).

Figura 3. Representación del número de copias del cromosoma 15 por array de mapeo genético. Los resultados se muestran para la relación log 2 (una medida del número de copias cromosómicas; eje Y) frente a la posición genómica en el cromosoma 15 (línea azul, número de copias 2 y cuadro rojo, deleción de una copia; eje X).

Tabla 2. Genes involucrados en la deleción del cromosoma 15 en anillo en la paciente.
Entre los 29 genes perdidos en la región de 15q26.3 se evidenció una interacción entre nueve de sus productos proteicos según el análisis por STRING (Figura 4). Los otros 20 genes tienen acciones individuales y no presentaron interacciones.

Figura 4. Interactoma de proteínas producto de los genes localizados en la región delecionada del cromosoma 15q26. Líneas celestes, interacciones determinadas en bases de datos seleccionadas; líneas rosadas, interacciones determinadas experimentalmente; líneas de otros colores, interacciones previstas por vecindad de genes, fusiones de genes y homología de proteínas.
DISCUSIÓN
Los componentes de la ancestría genética de la paciente es similar a la paciente que describimos anteriormente con cromosoma 15 en anillo 6 y conforme a la composición étnica del individuo ecuatoriano promedio 10. Se ha informado en muchos casos de la literatura, que el componente genético de ancestría podría determinar prevalencias diferentes según las regiones del mundo. Aunque no es definitivo, la presencia de dos casos en población ecuatoriana es llamativa 6.
Se han descrito diferentes mecanismos de formación de los cromosomas en anillo (1-4); nuestra paciente presenta un anillo producto de la deleción terminal de los brazos cortos y largos del cromosoma 15 1.
Las características clínicas de los casos con anillo del cromosoma 15 demuestran una gran variabilidad del fenotipo, es así que comparamos el de la actual paciente con el de los cien casos informados en la literatura 6-8. Encontramos un total de 52 rasgos, de los cuales el más común es el retraso del crecimiento, presente en el 75% del total de casos con cromosoma 15 en anillo, nuestra paciente también mostró esta característica. La frecuencia de los otros rasgos se observan en la Tabla 1: bajo peso al nacer, microcefalia, retraso mental, cara triangular, clinodactilia, braquimesofalangia, retraso en el desarrollo, trastorno del lenguaje, talla corta al nacer, manchas café con leche, puente nasal ancho, paladar ojival, edad ósea retrasada, retraso psicomotor, hipertelorismo ocular, orejas de implantación baja, micrognatia, manos pequeñas, filtrum corto, boca con comisuras dobladas hacia abajo, retrognatia, cuello corto, sobrepeso, hipotonía, pliegues epicánticos, problemas de alimentación, frente prominente, crecimiento desproporcionado de las extremidades, estrabismo, nariz pequeña, hernia diafragmática, cabello escaso, fisura palpebral corta, escoliosis, hipoplasia mamaria, comunicación interauricular, aracnodactilia, hipoplasia ovárica, genitales hipoplásicos, braquicefalia, hipoplasia de labios, pliegue palmar único, conducto arterioso persistente, defecto septal ventricular, pecho en escudo, falange dismórfica, pliegue palmar anormal en los dedos, hipoplasia uterina, comportamiento agresivo, hallux varus y discriminación visual.
Nuestra paciente presentó solo 12 de estas 52 características: retraso del crecimiento, bajo peso al nacer, microcefalia, retraso mental, retraso psicomotor y del desarrollo, hipertelorismo ocular, orejas de implantación baja, filtrum corto, labios hipolásicos, cuello corto y nariz pequeña. El fenotipo de la paciente mostró adicionalmente frente pequeña, arcos superciliares prominentes, orejas grandes, comisura labial abierta, hipertelorismo mamario y pectus excavatum.
Como diagnóstico diferencial, al no haber un fenotipo clásico de los pacientes con anillo del cromosoma 15, solamente se puede referir a síndromes polimalformativos o de fenotipo inespecífico no caracterizado. El fenotipo de los pacientes con anillos en general y del cromosoma 15 en particular, es muy variado y no es estándar. Se los puede agrupar como dismorfismos en estudio, posiblemente por cromosomopatías.
Los anillos dicéntricos o multicéntricos, anillos grandes o pequeños, como se muestran en la Figura 2, no están contemplados en el cariotipo de la paciente como líneas celulares, por estar presentes en un porcentaje menor al 3%, que es lo que al menos deberían superar para ser considerados como una línea celular. Son solamente variaciones del propio anillo que se forman por la dinámica de este tipo de alteraciones cromosómicas (rotura-reunión), y que antiguamente generaron discusiones científicas porque se pensaba que algo tenían que ver con el fenotipo. Este tipo de dinamismo del anillo, se lo puede observar y es gráficamente llamativo, pero no se asocia al fenotipo final 2, 6, 17.
La diversidad de características clínicas descritas en los casos con cromosoma 15 en anillo podría asociarse a la cantidad de material genético delecionado. Es frecuente encontrar el punto de rotura en 15q26 aunque se ha identificado en distintas bandas y sub-bandas del brazo largo del cromosoma 15.
Para determinar la longitud de la pérdida en el cromosoma 15 se ha empleado los arrays de mapeo genético, con los cuales no solo se ha definido el tamaño de la pérdida, sino que en algunos casos se ha identificado microduplicaciones que han estado asociadas a características clínicas consistentes con la duplicación de 15q26 como problemas cardíacos y renales 6, 7.
En nuestra paciente, la región delecionada de 3.459.037 pb de 15q26.3 contiene 29 genes. Entre ellos sobresalen: el receptor del factor de crecimiento similar a la insulina 1 (IGF1R). La haploinsuficiencia de este gen por deleción de 15q26 puede causar retraso del crecimiento pre y postnatal, situación descrita en 1991 y confirmada recientemente con el empleo de varias metodologías entre ellas: citogenética convencional, arrays de mapeo genético y secuenciación completa del exoma 12. CHSY1, este gen codifica un miembro de la familia de la condroitina N-acetilgalactosaminiltransferasa. Estas enzimas poseen actividad dual glucuroniltransferasa y galactosaminiltransferasa y desempeñan papeles críticos en la biosíntesis del sulfato de condroitina, un glicosaminoglicano involucrado en muchos procesos biológicos que incluyen la proliferación celular y morfogénesis. La expresión disminuida de este gen puede desempeñar un papel en el cáncer colorrectal, y las mutaciones en este gen son una causa del síndrome de braquidactilia preaxial a través de la modulación de la señalización NOTCH 13.
Otros genes, ubicados en la región delecionada, son PGPEP1L y TTC23 descritos frecuentemente en el llamado Síndrome de deleción del cromosoma 15q26-qter también conocido como Síndrome de deleción telomérica 15q, caracterizado por presentar varios síntomas entre ellos el más frecuente, el retraso del crecimiento pre y postnatal 14.
Al analizar los productos proteicos de los genes localizados en la zona de deleción en 15q26.3, se evidenció interacciones en 9 proteínas y de estas relaciones destaca IGF1R con CILP e IRS4. CILP, proteína de la capa intermedia del cartílago, que aumenta en el cartílago de la osteoartrosis temprana. Este tipo de artritis guarda una relación notable con la edad y es mucho más frecuente en mujeres que en varones. Tiene la capacidad de suprimir la proliferación inducida por IGF1 y la síntesis de proteoglicanos sulfatados, e inhibe la autofosforilación de IGF1R inducida por ligando (15). IRS4, actúa como una interfaz entre múltiples receptores de factores de crecimiento que poseen actividad tirosina quinasa, como el receptor de insulina, IGF1R y FGFR1, y una red compleja de moléculas de señalización intracelular que contienen dominios SH2 16.
Aunque existe una pérdida de material genético, y eso implica la disminución de funciones proteicas importantes, no encontramos una red de interacción precisa. Muchas proteínas actúan solas. Adicionalmente, la pérdida es en heterocigosis, por tanto, el alelo perdido en la deleción cromosómica, podría ser compensado en función con el alelo sobrante, esto podría explicar la alta variación de los fenotipos 12.
La pérdida de 3,5 Mb de 15q26.3 afecta la producción de proteínas eficaces, y el estudio de las redes de interacción de las proteínas alteradas, por esta haploinsuficiencia, evidencia el funcionamiento de cada gen dentro de un proceso biológico, que en este caso se tratan de interacciones establecidas en base a datos seleccionados y a determinados experimentalmente 12.
La variabilidad clínica observada en nuestra paciente y en todos los casos informados respondería al tamaño de la pérdida de ADN del cromosoma 15 involucrado en el anillo. Al igual que hemos analizado en otros casos de cromosomas en anillo 2, 6, 17, la heterogeneidad clínica y la frecuencia baja de la mayoría de características nos lleva a pensar que es muy difícil describir un fenotipo estándar y conceptualizar un Síndrome del anillo 18, nos reafirmamos que el fenotipo alterado está definido principalmente por la cantidad de material genético involucrado en la formación del anillo y la pérdida de los genes de la región específica.
CONCLUCIONES
La utilización de herramientas citogenéticas y moleculares convencionales ha permitido el progreso del diagnóstico genético y una mejor comprensión de los síndromes genéticos. Todos los reordenamientos por los que pasa un cromosoma para adquirir una estructura de anillo presentan un patrón similar. Comienza con una rotura de ambos segmentos terminales con la posterior unión de los extremos rotos. Sin embargo, el fenotipo de un paciente con un cromosoma 15 en anillo es muy variable y no hay mucha información disponible sobre la pérdida de material genético en estos casos. Aunque se ha informado desde 1966 casos similares al presente, solamente un par de ellos hacen referencia a la cantidad de pérdida del material genético 6, 7, 8. Este caso clínico es un nuevo aporte a la comprensión científica de los anillos cromosómicos y es el segundo reporte de un paciente ecuatoriano con un cromosoma 15 en anillo, aplicando herramientas citogenéticas y moleculares
REFERENCES
1. Guilherme RS, Meloni VFA, Kim CA, Pellegrino R, Takeno SS, Spinner NB, Conlin LK, Christofolini DM, Kulikowski LD, Melaragno MI. Mechanisms of ring chromosome formation, ring instability and clinical consequences. BMC Med Genet. 2011;12:171–7.
2. Paz-y-Miño C, Proaño A, Verdezoto S, García JL, Hernández-Rivas JM, Leone PE. Clinical, cytogenetic, and molecular findings in a patient with ring chromosome 4: case report and literature review. BMC Med Genomics.2019;12(1):167.
3. Côté GB, Katsantoni A, Deligeorgis D. The cytogenetic and clinical implications of a ring chromosome 2. Ann Genet. 1981;24:231–5.
4. Sigurdardottir S, Goodman BK, Rutberg J, Thomas GH, Jabs EW, Geraghty MT. Clinical, cytogenetic and fluorescence in situ hybridization findings in two cases of “complete ring” syndrome. Am J Med Genet. 1999;87:384–90.
5. Butler MG, Fogo AB, Fuchs DA, et al. Two patients with ring chromosome 15 syndrome. Am J Med Genet. 1988;29:149-54.
6. Paz-y-Miño C, Guevara-Aguirre J, Paz-y-Miño A, Velarde F, Armendáriz-Castillo I, Yumiceba V, Hernández JM, García JL, Leone PE. Ring chromosome 15 - cytogenetics and mapping arrays: a case report and review of the literature. J Med Case Rep. 2018;12(1):340-9.
7. Shao HY, Wang HL, Wu H, Liu XY, Miao ZY. Clinical, cytogenetic and molecular analyses of a rare case with ring chromosome 15 and review of the literature. Taiwan J Obstet Gynecol. 2020;59(6):980-4.
8. Zhang J, Yang Y, Zhang J, Wang S, Yao F, Zhang Y, Jiang S. Molecular cytogenetic study of a case with ring chromosome 15. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2021;38(3):238-41 [En Chino].
9. Liu YH, Chang SD, Chen FP. Increased fetal nuchal fold leading to prenatal diagnosis of ring chromosome 15. Prenat Diagn. 2001;21(12):1031-3.
10. Zambrano AK, Gaviria A, Cobos-Navarrete S, Gruezo C, Rodríguez-Pollit C, Armendáriz-Castillo I, García-Cárdenas JM, Guerrero S, López-Cortés A, Leone PE, Pérez-Villa A, Guevara-Ramírez P, Yumiceba,V, Fiallos G, Vela M, Paz-y-Miño C. The three-hybrid genetic composition of an Ecuadorian population using AIMs-InDels compared with autosomes, mitochondrial DNA and Y chromosome data. Sci Rep. 2019;9(1):9247.
11. STRING CONSORTIUM 2021. [Internet]. [Consultado 2 May 2021]. Disponible en: https://string-db.org/cgi/input?sessionId=bREgW3EM4iOP&input_page_show_search=on.
12. Ocaranza P, Golekoh MC, Andrew SF, Guo MH, Kaplowitz P, Saal H, Rosenfeld RG, Dauber A, Cassorla F, Backeljauw PF, Hwa V. Expanding genetic and functional diagnosis of IGF1R haploinsufficiencies. Horm Res Paediatr. 2017;87:412-22.
13. Tian J, Ling L, Shboul M, Lee H, O´Connor B, Merriman B, Nelson SF, Cool S, Ababneh OH, Al-Hadidy A, Masri A, Hamamy H, Reversade B. Loss of CHSY1, a secreted FRINGE enzyme, causes syndromic brachydactyly in humans via increased NOTCH signaling. Am J Hum Genet. 2010;87(6):768-78.
14. MalaCards. Human Disease Database: Chromosome 15q26-qter Deletion syndrome. 2021. [Internet]. [Consultado Jun 2021]. Disponible en: https://www.malacards.org/card/chromosome_15q26_qter_deletion_syndrome.
15. Liu L, He J, Liu C, Yang M, Fu J, Yi J, Ai X, Miao Liu M, Yong Zhuang Y, Zhang Y, Huang B, Li C, Zhou Y, Feng C. Cartilage intermediate layer protein affects the progression of intervertebral disc degeneration by regulating the extracellular microenvironment (Review). Int J Mol Med. 2021;47(2):475-84.
16. Heinen CA, de Vries EM, Alders M, Bikker H, Zwaveling-Soonawala N, van den Akker ELT, Bakker B, Hoorweg-Nijman G, Roelfsema F, Hennekam RC, Boelen A, van Trotsenburg ASP, Fliers E. Mutations in IRS4 are associated with central hypothyroidism. J Med Genet. 2018;55(10):693-700.
17. Paz-y-Miño C, Benítez J, Ayuso C, Sanchez-Cascos A. Ring chromosome 6: clinical and cytogenetic behaviour. Am J Med Genet. 1990;35:481-3.
18. Kosztolányi G. Does “ring syndrome” exist? An analysis of 207 case reports on patients with a ring autosome. Hum Genet. 1987;75:174-9.
Received: 30 June / Accepted: 7 November 2021 / Published: date. 15 February 2022
Citation: Paz-y-Miño C, Medranda C, Loaiza A, Ponce M, Leone PE. Rara patología derivada de un anillo del cromosoma 15. Clínica, genómica e interactoma de proteínas de genes asociados al fenotipo. Revis Bionatura 2022;7(1). 16. http://dx.doi.org/10.21931/RB/2022.07.01.16