2021.06.03.32
Files > Volume 6 > Vol 6 No 3 2021
Metachromatic Leukodystrophy: Diagnosis and Treatment Challenges

Nayibe Tatiana Sanchez-Alvarez 1.2.3; Paula Katherine Bautista-Niño 2; Juanita Trejos-Suárez 3, Norma Cecilia Serrano-Diaz2
Available from: http://dx.doi.org/10.21931/RB/2021.06.03.32
SUMMARY
Metachromatic leukodystrophy is a neurological disease of the lysosomal deposit that has a significant impact given the implications for the neurodegenerative deterioration of the patient. Currently, there is no treatment available that reverses the development of characteristic neurological and systemic symptoms. Objective. Carry out an updated bibliographic search on the most critical advances in the treatment and diagnosis for LDM. A retrospective topic review published in English and Spanish in the Orphanet and Pubmed databases. Current treatment options, such as enzyme replacement therapy and hematopoietic stem cell transplantation aimed at decreasing the rapid progression of the disease, improving patient survival; however, these are costly. The pathophysiological events of intracellular signaling related to the deficiency of the enzyme Arylsulfatase A and subsequent accumulation of sulphatides and glycosylated ceramides have not yet been established. Recently, the accumulation of C16 sulphatides has been shown to inhibit glycolysis and insulin secretion in pancreatic cells. The significant advance in technology has allowed timely diagnosis in patients suffering from LDM; however, they still do not have an effective treatment.
Keywords: Metachromatic leukodystrophy, treatment, diagnosis, prevalence.
INTRODUCTION
Metachromatic leukodystrophy (LDM) is a neurodegenerative lysosomal deposition disease caused by a deficiency in the enzyme Arylsulfatase A (ARSA), which leads to an accumulation of sulfates in lysosomes, lipids of great importance in the structure of myelin. The lysosomal deposition of sulphatides in neurons and myelinating cells produces severe demyelination and neurodegeneration of central (CNS) and peripheral nervous systems (PNS) 1,2.
According to the progress of the disease and the onset of symptoms, it is classified into three stages: late infantile, juvenile, and adult 3. Although there is still no effective treatment to reverse the symptoms, different therapeutic strategies are in the research and deepening phase3.
There are no pathological mechanisms by which the accumulation of sulfates leads to lamyelination in CNS and PNS cells. However, in in vitro and in vivo models of pancreatic B cells, the accumulation of sulphatides can inhibit insulin secretion and therefore disable the glycolytic function of the cells 4–6. The main objective of this review is to carry out an updated bibliographic search on the most critical advances in the treatment and diagnosis of LDM.
METHODOLOGY
Retrospective review article consisted of searching for articles published in English and Spanish in the Orphanet and Pubmed databases with the keywords, Medical Subject Headings (MeSH): Metachromatic leukodystrophy, treatment, diagnosis, prevalence.
Review articles, meta-analyses, systematic reviews, and studies that preferably had descriptions of cellular mechanisms in LDM were included; 80 relevant references are included due to their historical antecedents considered valuable to complement the information and have LDM criteria. Other references support the descriptions of essential mechanisms in this pathology.
RESULTS
3086 articles containing MeSH were found: When specifying "Metachromatic leukodystrophy", 1,650 articles were found, 410 with the words "Metachromatic leukodystrophy, treatment," 932 with "Metachromatic leukodystrophy, diagnosis" and 94 "Metachromatic leukodystrophy, prevalence". Of all the articles, 80 references were located considering that metachromatic leukodystrophy is the infantile stage and studies in which new diagnostic technologies and treatments were mentioned, Figure 1.
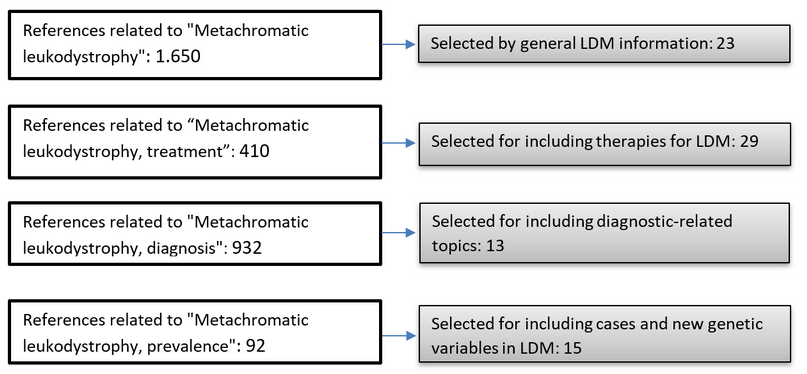
Figure 1. List of findings and selection of references for this review article.
LEUKODYSTROPHIES
According to the National Institute of Health of the United States (NIH) and the Neurological Disorders Unit, leukodystrophies or leukoencephalopathies 7 are a group of rare diseases caused by genetic defects, leading to the presence of chemical deposits within cells of both the CNS and the PNS, resulting in the deterioration of the myelin sheath 8,9.
This pathology is characterized by demyelinating processes that primarily affect the CNS and later the PNS 10,11. Leukodystrophies have an incidence of 1 in 7,500 live births; however, only half of the patients receive a specific diagnosis 7,12.
The European Association of Leukodystrophies (ELA) classification includes those caused by peroxisomal content, lysosome, vacuolation, undetermined atypical hypomyelination. All of these leukodystrophies target myelin; in some cases, its degradation occurs, and in others, there is no formation of it 13,14.
Specific leukodystrophies include metachromatic leukodystrophy, Krabbé disease, adrenoleukodystrophy, Pelizaeus-Merzbacher disease, Canavan disease, and childhood ataxia hypomyelination of the central nervous system (also known as substance disappearance disease or white matter), Alexander's disease, Refsum's disease, and cerebrotendinous xanthomatosis 15.
The progress of the disease is rapid and devastating. It can occur at any age, both in childhood and in adults 12. Currently, few reports are generated on diagnostic tests, treatment, and the course of this disease. The current diagnosis is based on clinical history, examinations, radiological and laboratory findings, including genetic tests 12,15. This review summarizes the knowledge about the epidemiology, pathophysiology, diagnosis, and treatment of Metachromatic Leukodystrophy.
Metachromatic Leukodystrophy (MLD)
MLD was first described in 1910 by Perisini and Alzheimer's in adult patients 3,16. In 1925, Scholz published a pathological clinical study of juvenile MLD, and 34 years later, Peiffer showed that the neural tissues in Scholz's study had metachromatically stained sections. In 1958, independently, Jatzkewitz discovered that the metachromatic sections resulted from the accumulation of sulphatides. On the other hand, Austin and his colleagues were the ones who discovered the defect in the activity of the enzyme arylsulfatase A. Greenfield made the first report of infantile MLD in 1933, and the term "metachromatic leukodystrophy" (metachromatischen Leukodystrophien) was first used by Peiffer in 1959 to describe what was previously known as "diffuse cerebral sclerosis" 16,17.
MLD (OMIM # 250100) is a lysosomal storage disorder with an autosomal recessive pattern of inheritance, so carriers of one copy of the abnormal gene are not affected by the disease 18,19. MLD is caused by mutations in the ARSA gene, located on chromosome 22q13.33 20, that encodes the lysosomal enzyme Arylsulfatase A (ARSA) (ARSA; OMIM 607574, GenBank accession number, NG_009260) 21,22.
Among the types of mutations that have been reported in ARSA are amino acid substitutions, nonsense mutations, deletions, and sense-change mutation. ARSA is a crucial enzyme in the catabolism of sphingolipids, an abundant component of myelin, Figure 2. The progressive accumulation of sulphatides in the lysosomes of neurons and myelinating cells (Schwann cells in the PNS, oligodendrocytes in the CNS) causes severe demyelination and neurodegeneration progressive neurological deficits, and premature death in childhood form 23.
MLD has three clinical subtypes: late-infantile, juvenile, and adult 17. Each of the subtypes has characteristics that make them different from each other.
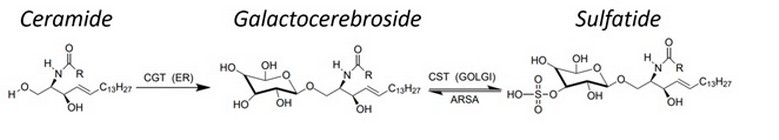
Figure 2. Synthetic sulfatide route
Late childhood MLD: The start occurs before 30 months. Typical findings are weakness, hypotonia, clumsiness, frequent falls, difficulty walking, and dysarthria. As the disease progresses, motor, cognitive, and language skills deteriorate. Later signs include spasticity, pain, seizures, impaired vision, and hearing. Of the cases of MLD, 50 to 60% are of late childhood presentation 24. In the final stages, children have tonic spasms, mindless posture, and general ignorance of their environment 17.
Juvenile MLD: The clinical signs of this pathology appear between 30 months and 16 years of age. This clinical form of the disease represents 20 to 30% of MLD cases 25. Initial manifestations include decreased school performance and the appearance of behavioral problems, followed by gait disturbances. Progression is similar but slower than in the late-infant form 17.
Adult MLD: Symptom presentation occurs after 16, sometimes up to the fourth or fifth decade, and represents 10 - 20% of the MLD cases 25. Initial signs include problems with school or work performance, personality changes, emotional lability, or psychosis; in other patients, neurological symptoms (weakness and loss of coordination that progresses to spasticity and incontinence) or seizures predominate. Peripheral neuropathy is common. The course of the disease is variable, with periods of stability interspersed with periods of decline. The final stage is similar to the juvenile and infantile forms 17.
Epidemiology - Prevalence
Currently, there are no relevant reports of prevalence worldwide, since there is talk of a set of orphan llama diseases, considered in the countries of the European Union as one that affects one in 2,000 people; less than 200,000 people affect the United States, less than 50,000 people in Japan, less than one in 10,000 in Taiwan and one in 5,000 in Colombia 26.
However, in the Orphanet database that describes the MLD, an estimated prevalence of 1 case in 625,000, with an incidence of 0.5 and 1 in 50,000 25,27–30.
In Latin America, the reports generated so far are focused on describing cases and mutations in the ARSA gene 31–37.
PATHOPHYSIOLOGICAL MECHANISMS OF THE DISEASE
MLD is a neurodegenerative disease characterized by the accumulation of sulfates (sulfated glycosphingolipids, especially sulfogalactocerebrosides or sulfogalactosylceramides) in the central nervous system, kidneys, and in other organs such as the retina, liver, testes, pancreas, sweat glands, adrenal cortex, and tissue. rectal 38. The PNS is systematically affected, causing a decrease in nerve conduction velocities. This disorder progresses to a state of decerebration after a few years, resulting in death within 5 years after the onset of symptoms 39.
Galactosylceramide (GalC) and its sulfated form, the sulphatides, are two glycosphingolipids found in high concentrations in the CNS and PNS cell membranes. GalC and sulphatides comprise 23% and 4% of the total mass of myelin lipids, respectively, and together account for about a third of the content of the myelin sheath40,41. Among the primary myelinating cells, oligodendrocytes and sulfate-producing Schwann cells 42. In the nervous system, sulphatides are related to intercellular recognition, cell differentiation, the interaction of the myelinating cell with the axon, the maintenance of the axonal structure, and nerve conduction 39, 41, 42.
The myelin sheath is an extended membrane, which spirally wraps around a portion of the axon. Each myelin sheath is extended by modifying the cell membrane of oligodendrocytes and Schwann cells, forming a structure with several myelin segments surrounding the axon (Figure 3) 40.
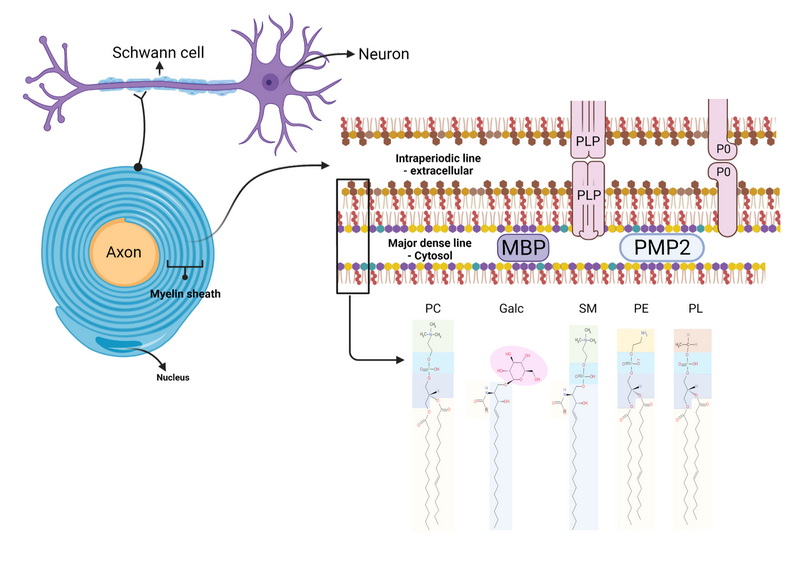
Figure 3. Structure of the myelin sheath
Source: Figure adapted from Poitelon Y. et al., 2020 26.
Diagram of the structure of the myelin sheath, (I) myelinated axon, (II) myelin sheath, (III) Bilayer membrane, and (IV) Lipid classes. Myelin is formed by the opposition of the external surfaces and the internal surfaces of the myelin bilayer that constitute the intraperiodic line and the dense main line, respectively (II, III). The myelin bilayer has an asymmetric lipid composition (III, IV). The myelin protein is also distributed asymmetrically, such as PLP (Proteolipid Protein) and P0 in the intraperiodic line and the dense mainline, the PMP2 protein in the dense main line (III), as well as the composition of cholesterol, phospholipids (e.g., Plasmogen, lecithin, sphingomyelin), glycolipids (eg, Galactosylceramide) and other phospholipids. Proteins P0, PMP2, and enrichment of sphingomyelin in myelin are specific for myelin PNS.
Each oligodendrocyte can contribute to the protection of around 50 different axons. In addition to its importance in driving the nerve impulse, myelin may have a symbiotic relationship with the axon 45. Other publications suggest that the development of myelin occurs synergistically with the axon and that the axonal cytoskeleton does not form correctly in the absence of myelin 46. Myelin is usually synthesized in MLD. However, the progressive accumulation of sulfatides induces instability of the myelin membrane, leading to final demyelination.
Myelin formation occurs in two ways. In the CNS, widely branched oligodendrocytes form myelin around multiple axons at the same time, allowing for higher neuronal density in the brain. On the other hand, in the PNS, Schwann cells circularize an axon with several layers of myelin, forming a multilayer structure. Although the primary function of myelin is the same in the CNS and PNS, the molecular requirements for myelin formation differ. In general terms, myelination is a complex process during which a set of proteins and lipids are synthesized in a coordinated way by glial cells (Schwann cells and Oligodendrocytes). i) the pluripotent precursors of Schwann and Oligodendrocyte cells must differentiate; for this, a set of transcription factors is involved in the cell's potential development and its myelinating function. ii) the precursor cells must migrate to the myelination site; this step is believed to require communication between the migrant cells and the axons. iii) when the cell contacts the axon, transcriptional and post-transcriptional changes force the exit of the cell cycle and the differentiation of the cell into a mature Oligodendrocyte or Schwann cell. iv) Myelin synthesis is carried out around the axon to form a compact and functional structure to finally v) ensure that the structure and function of myelin are preserved through the continuous expression and synthesis of each of the components of myelin 46.
Although many of the myelin components have been related to the assembly and maintenance of the structure of this 47, Galactolipids and sulfatides, the main components of myelin, have received significant attention. The synthesis and maturation of most of these compounds begin in the endoplasmic reticulum and rapidly spreads to the extracellular space through the early secretion system 48,49.
As already mentioned, this myelination process is continuous, establishing a balance in the amount of each of its compounds.
Diverse studies 50–52 have shown that mitochondria and lysosomes are critical organelles for the maintenance of homeostasis in myelinating cells; likewise, there is a close relationship between these two and the interaction directly with cellular stress 50, In this way, communication between mitochondria and lysosomes can lead to mitophagy, processes in which damaged mitochondria target autophagosomes for lysis. Autophagosomes subsequently fuse with lysosomes / late endosomes to generate autolysosomes that mediate the degradation of mitochondria, or in contrast, mitochondria and lysosomes can also interact directly through nondegradable processes through the dynamic formation of sites of Membrane contact between organelles in healthy mammalian cells, leading to transfer of lipid, calcium, and iron metabolites, regulation of organelle dynamics, such as mitochondrial division and endosomal division 50.
However, the misregulation of mitochondria or contacts with lysosomes can simultaneously lead to the dysfunction of both organelles in various lysosomal storage disorders (LSD) such as MLD, which has been genetically and functionally related to mitochondrial and lysosomal defects 53.
DIAGNOSIS
Diagnosis of MLD requires a high level of complexity; however, Technological development is allowing the evolution of new diagnostic strategies aimed not only at identifying ARSA enzyme deficiency but also at the presence of findings such as progressive neurological dysfunction or evidence of typical lesions on magnetic resonance imaging, characteristic of the disease.
For the diagnosis of MLD, one of the following events must be present:
- Identification of biallelic ARSA pathogenic variants in molecular genetic tests (see Table 1)
- Identification of increased urinary excretion of sulfatides
- Identification of metachromatic lipid deposits in a nerve or brain biopsy
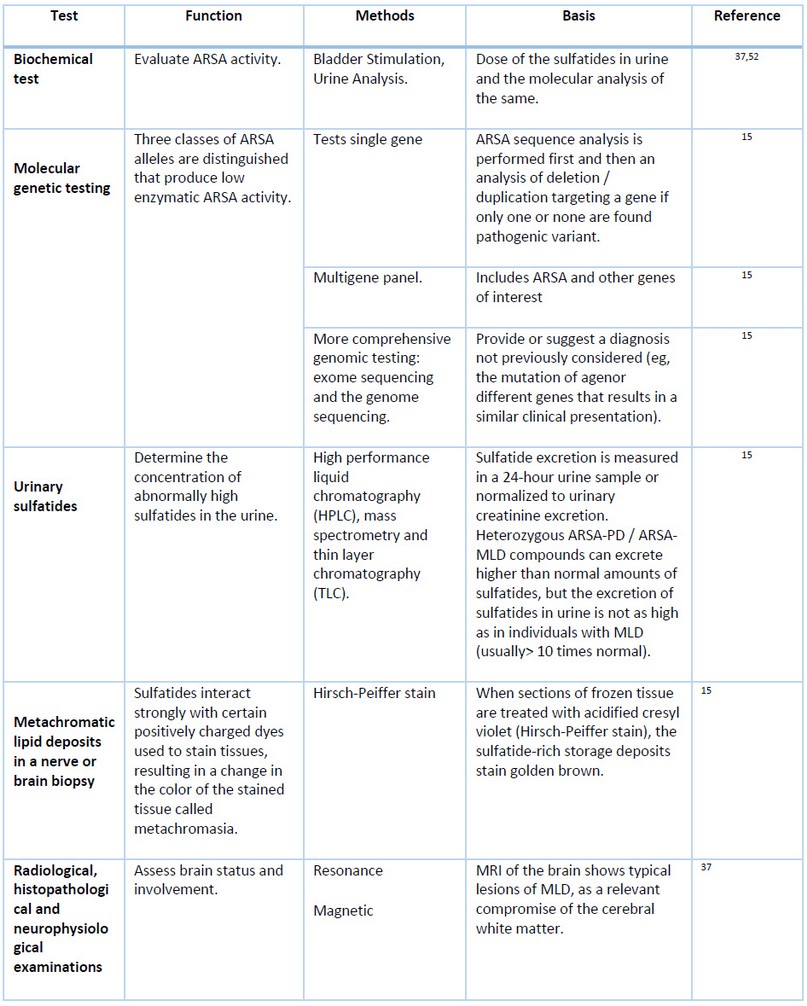
Table 1. Diagnostic methods for the identification of MLD.
THERAPIES - TREATMENT
Due to the lack of information on this disease, few treatments aimed at safeguarding the lives of patients, mainly because the barrier between the blood and the brain limits the access of the recombinant product to the nervous tissues 54,55.
Currently, there is no treatment available to reverse the fatal outcome of this devastating disease; therapy is only supportive, disease severity and lack of effective therapies describe the need for innovative therapeutic approaches 54, however, according to the database of clinicaltrials.gov, To date, 36 clinical trials have been registered, of which 6 are in an active state, focused on determining the natural history of the disease, efficacy, and safety of HGT-1110 (recombinant human arylsulfatase A), gene therapy based on lentiviral vectors, biomarker search, Human placental-derived stem cell transplantation in participants with MLD.
Symptom treatment and support.
At the beginning of the disease, the patient is treated with remedies for epilepsy, muscle relaxants for contractures, physical therapy to improve nerve and muscle function, stimulation to maximize the intellect, and finally support to the families of those affected so that the parents and/or caregivers can anticipate decisions about wheelchairs, feeding tubes, and other care 9,36,56.
Enzyme replacement therapy
Enzyme replacement therapy has become the most promising therapeutic option for various lysosomal diseases due to its potential to compensate for the deficiency; it is currently available as a treatment for some disorders such as Fabry, Gaucher, Pompe, mucopolysaccharidosis I, II, VI and it's under development for others like MLD 54. However, this type of therapy remains challenging because the blood-brain barrier is impervious to lysosomal enzymes 57,58.
Currently, there are three-phase I and II studies reporting the safety, efficacy, and pharmacodynamics of this type of therapy in patients with MLD 59–61. In this therapy, recombinant human arylsulfatase A (rhASA) has been used, which has generated changes in the concentrations of sulfates in CSF and urine of the patients from the beginning to the end of the study, demonstrating an excellent therapeutic start 61,62.
Gene therapy
Therapy based on the overexpression of wild-type ARSA in different cell types. This type of therapy has been successful when autologous hematopoietic stem cells obtained from patients with MLD are used, in which the ARSA gene is overexpressed by gene transfer using retroviral or lentiviral vectors 63. The wild-type synthesized ARSA secreted by these genetically engineered autologous hematopoietic stem cells are absorbed by adjacent ARSA-deficient cells through the mannose-6-phosphate receptor system 64.
Today, there are complete and ongoing clinical trials in which gene therapy has been used as an adjunct in patients with MLD 65.
Hematopoietic stem cell therapy
With this type of therapy, it is possible to reach the blood-brain barrier by using hematopoietic stem cells that differentiate into microglial cells 66. In principle, once the donor's microglial cells cross the blood-brain barrier, they can secrete the wild-type ARSA enzyme, taken up by ARSA-deficient receptor neuronal cells. This uptake is performed by the mannose-6-phosphate receptor system, which carries the enzyme directly to the lysosomes, where the sulfatides accumulate in the ARSA receptor-deficient neural cells 63,67.
Despite being a promising therapy in clinical trials, it has been shown that the main complication that can occur in treated patients is the presence of graft-versus-host disease. The drugs used for this type of therapy have been busulfan, cyclophosphamide, anti-thymocyte globulin (ATG), tested on both children and adults 67,68.
Brain gene therapy with adeno-associated virus (AAV) vectors
This type of therapy is ideal for treating MLD patients since the genetic message for the ARSA gene could be sent directly to the SNC. This therapy has the use of AAV vectors that have low immunogenicity, low oncogenic potential through insertion mutagenesis, and in addition to this, three main characteristics that make it unique:
- Because it is safe, it can be administered directly in the CNS to Through the intraparenchymal, intracerebroventricular, and intracisternal routes where the ARSA protein is expressed persistently and permanently in a single dose.
- Once this therapy is administered at the site of action, it has the ability to prevent the symptoms of the disease 69–71.
- The ARSA protein when expressed in healthy cells is capable of cross-correcting neighboring cells, therefore cells that are corrected will secrete the protein that is endocitated through the mannose-6-phosphate receptor (M6P) pathway 72.
AAVs that have been used so far for the treatment of MLD have been AAV5, and then with the discovery of serotypes derived from non-human primates, studies with AAVrh were conducted 71,73–75.
CRISPR Cas9
This technique has become a novel and widely used research tool with which rapid, easy and efficient genetic modification is achieved 76, with which alleles that cause disease are repaired by changing the DNA sequence at the exact location of the chromosome 10. The acronym CRISPR Cas9 comes from Clustered Regularly Interspaced Short Palindromic Repeats, in Spanish "Short Palindromic Repeats Grouped and Regularly interspaced".
This technology has enabled two-stage genome editing. In the first, guide RNA, which is specific to a DNA sequence, is associated with the enzyme Cas9 endonuclease, which works by cutting DNA. In the second stage, natural repair mechanisms of the cut DNA are activated. In which indel (insertion-deletion) can be generated or, moreover, a specific sequence is incorporated exactly at the original cut site. For this, the sequence of interest is supplied to the cell in such a way that it is integrated into the DNA 77.
This technology brings hope of a cure not only for MLD but for many other genetic diseases such as Alzheimer's, Cancer, AIDS, Cystic Fibrosis, etc., for many genetic diseases. It should be noted that to date it has not been used as a treatment for patients with this type of disease 78.
CONCLUSIONS
Despite great research efforts to find an effective treatment for MLD, these approaches have not been effective not only because of the low efficiency of gene editing but because gene therapies must cross the blood-brain barrier. which becomes a challenge. Therefore, patients with MLD continue without any treatment and, above all, there are still gaps in knowledge about the management of this disease.
Given the limitations presented by existing therapies in the treatment of MLD, it is necessary to investigate new therapies to increase the effectiveness of the treatment.
Considering the panorama in MLD, it is essential to continue in the constant search that points to the understanding of epidemiology and diagnostic tools for the disease.
Financial support: This project was funded by the project entitled "Metabolism of sulfatides, glycolysis and mitochondrial function in metachromatic leukodystrophy" in the call for research projects in basic sciences 712-2015, of Minciencias. Whose principal investigator is Dr. Norma C. Serrano.
Conflict of interests: None
Abbreviations
MLD: Metachromatic Leukodystrophy
CNS: Central Nervous System
PNS: Peripheral Nervous System
CRISPR Cas9: Short Palindromic Repetitions Grouped and Regularly Spaced
ATP: Adenosine triphosphate
GalC: Galactosylceramide
rhASA: Recombinant Human Arylsulfatase A
ATG: Anti-thymocyte Globulin
REFERENCES
1. Takahashi T, Suzuki T. Role of sulfatide in normal and pathological cells and tissues. J Lipid Res. 2012;53(8):1437-50.
2. Tan M, Fuller M, Zabidi-Hussin Z, Hopwood JJ, Meikle PJ. Biochemical profiling to predict disease severity in metachromatic leukodystrophy. Mol Genet Metab. 2010;99(2):142-8.
3. OMIM - Herencia mendeliana en línea en el hombre [Internet]. [Updated April 16, 2021, cited 13 of July de 2019]. available in: https://www.omim.org/
4. Blomqvist M, Carrier M, Andrews T, Pettersson K, Mansson JE, Rynmark BM, et al. In vivo administration of the C16:0 fatty acid isoform of sulfatide increases pancreatic sulfatide and enhances glucose-stimulated insulin secretion in Zucker fatty (fa/fa) rats. Diabetes Metab Res Rev. 2005;21(2):158-66.
5. Boslem E, Meikle PJ, Biden TJ. Roles of ceramide and sphingolipids in pancreatic β-cell function and dysfunction. Islets. 2012;4(3):177-87.
6. Buschard K, Blomqvist M, Månsson J-E, Fredman P, Juhl K, Gromada J. C16:0 Sulfatide Inhibits Insulin Secretion in Rat β-Cells by Reducing the Sensitivity of KATP Channels to ATP Inhibition. Diabetes. 2006;55(10):2826.
7. Gulati S, Jain P, Chakrabarty B, Kumar A, Gupta N, Kabra M. The spectrum of leukodystrophies in children: Experience at a tertiary care centre from North India. Ann Indian Acad Neurol. 2016;19(3):332-8.
8. Leucodistrofia : National Institute of Neurological Disorders and Stroke (NINDS) [Internet]. [Updated December 21, 2016, cited 12 de November of 2019]. available in: https://espanol.ninds.nih.gov/trastornos/leucodistrofia.htm
9. Leucodistrofia metacromática | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program [Internet]. [updated 10/6/2015, cited 31 de December of 2018]. available in: https://rarediseases.info.nih.gov/espanol/12627/leucodistrofia-metacromatica
10. Van der Knaap MS, Wolf NI, Heine VM. Leukodystrophies: Five new things. Neurol Clin Pract. 2016;6(6):506-14.
11. Beerepoot S, Nierkens S, Boelens JJ, Lindemans C, Bugiani M, Wolf NI. Peripheral neuropathy in metachromatic leukodystrophy: current status and future perspective. Orphanet J Rare Dis. 2019
12. Gordon H, Letsou A, Bonkowsky J. The Leukodystrophies. Semin Neurol. 2014;34(03):312-20.
13. Association Européenne Contre les Leucodystrophies. Accueil - [Internet]. [cited 15 of November of 2019]. available in: https://ela-asso.com/
14. Brignone MS, Lanciotti A, Camerini S, De Nuccio C, Petrucci TC, Visentin S, et al. MLC1 protein: a likely link between leukodystrophies and brain channelopathies. Front Cell Neurosci. 2015;9:66-66.
15. Gordon-Lipkin E, Fatemi A. Current Therapeutic Approaches in Leukodystrophies: A Review. J Child Neurol. 2018;33(13):861-8.
16. Nyhan WL, Barshop BA, Ozand PT. Atlas of Metabolic Diseases Second edition. CRC Press; 2005.
17. Gomez-Ospina N. Arylsulfatase A Deficiency. En: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editores. GeneReviews((R)). Seattle WA: University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle; 1993.
18. Almarzooqi S, Quadri A, Albawardi A. Gallbladder Polyps in Metachromatic Leukodystrophy. Fetal Pediatr Pathol. 2018;37(2):102-8.
19. Van den Broek BTA, Page K, Paviglianiti A, Hol J, Allewelt H, Volt F, et al. Early and late outcomes after cord blood transplantation for pediatric patients with inherited leukodystrophies. Blood Adv. 2018;2(1):49-60.
20. Golchin N, Hajjari M, Malamiri RA, Aminzadeh M, Mohammadi-asl J, Golchin N, et al. Identification of a novel mutation in ARSA gene in three patients of an Iranian family with metachromatic leukodystrophy disorder. Genet Mol Biol. 2017;40(4):759-62.
21. Belli G, Bartolini E, Bianchi A, Mascalchi M, Stagi S. Central Precocious Puberty in a Child With Metachromatic Leukodystrophy. Front Endocrinol. 2018;9.
22. Bindu P, Mahadevan A, Taly A, Christopher R, Gayathri N, Shankar S. Peripheral neuropathy in metachromatic leucodystrophy. A study of 40 cases from south India. J Neurol Neurosurg Psychiatry. 2005;76(12):1698-701.
23. Calbi V, Fumagalli F, Consiglieri G, Penati R, Acquati S, Redaelli D, et al. Use of Defibrotide to help prevent post-transplant endothelial injury in a genetically predisposed infant with metachromatic leukodystrophy undergoing hematopoietic stem cell gene therapy. Bone Marrow Transpl. 2018.
24. Wang RY, Bodamer OA, Watson MS, Wilcox WR. Lysosomal storage diseases: Diagnostic confirmation and management of presymptomatic individuals. Genet Med. 2011;13(5):457-84.
25. Dehghan Manshadi M, Kamalidehghan B, Aryani O, Khalili E, Dadgar S, Tondar M, et al. Four novel ARSA gene mutations with pathogenic impacts on metachromatic leukodystrophy: a bioinformatics approach to predict pathogenic mutations. Ther Clin Risk Manag. 2017;13:725-31.
26. Ministerio de Salud y Protección Social, Consuelo Pinzón Gutiérrez, Fredy Orlando Mendivelso Duarte, Sandra Patricia Misnaza Castrillón. HUÉRFANAS-RARAS code: 342 [Internet]. p. 19. available in: https://www.ins.gov.co/buscador-eventos/Lineamientos/PRO%20Enfermedades%20hu%C3%A9rfanas%20y%20raras.pdf#search=enfermedades%20raras
27. Prevalence of rare diseases: Bibliographic data [Internet]. [cited 28 of November of 2018]. available in: http://online.fliphtml5.com/trmf/ktss/#p=10
28. Kehrer C, Groeschel S, Kustermann-Kuhn B, Burger F, Kohler W, Kohlschutter A, et al. Language and cognition in children with metachromatic leukodystrophy: onset and natural course in a nationwide cohort. Orphanet J Rare Dis. 2014;9(18):1750-1172.
29. Krägeloh-Mann I, Groeschel S, Kehrer C, Opherk K, Nägele T, Handgretinger R, et al. Juvenile metachromatic leukodystrophy 10 years post transplant compared with a non-transplanted cohort. Bone Marrow Transplant. 2012;48:369.
30. Doherty K, Frazier SB, Clark M, Childers A, Pruthi S, Wenger DA, et al. A closer look at ARSA activity in a patient with metachromatic leukodystrophy. Mol Genet Metab Rep. 2019;19.
31. Artigalás O, Lagranha VL, Saraiva-Pereira ML, Burin MG, Lourenço CM, van der Linden H, et al. Clinical and biochemical study of 29 Brazilian patients with metachromatic leukodystrophy. J Inherit Metab Dis. 2010;33(S3):257-62.
32. Virgens MYF, Siebert M, Bock H, Burin M, Giugliani R, Saraiva-Pereira ML. Genotypic characterization of Brazilian patients with infantile and juvenile forms of metachromatic leukodystrophy. Gene. 2015;568(1):69-75.
33. Saute JAM, Souza CFM de, Poswar F de O, Donis KC, Campos LG, Deyl AVS, et al. Neurological outcomes after hematopoietic stem cell transplantation for cerebral X-linked adrenoleukodystrophy, late onset metachromatic leukodystrophy and Hurler syndrome. Arq Neuropsiquiatr. 2016;74(12):953-66.
34. Alvarez-Leal M, Contreras-Hernández D, Chávez A, Diaz-Contreras JA, Careaga-Olivares J, Zúñiga-Charles MA, et al. Leukocyte arylsulfatase A activity in patients with alcohol-related cirrhosis: Arylsulfatase A in Chronic Alcoholic Cirrhosis. Am J Hum Biol. 2001;13(3):297-300.
35. Echeverri Olga Y, Salazar Diego A, Rodriguez-Lopez A, Janneth G, Almeciga-Diaz Carlos J, Barrera Luis A. Understanding the Metabolic Consequences of Human Arylsulfatase A Deficiency through a Computational Systems Biology Study. Cent Nerv Syst Agents Med Chem. 2016.
36. Espejo LM, de la Espriella R, Hernández JF. Leucodistrofia metacromática. Presentación de caso. Rev Colomb Psiquiatr. 2017;46(1):44-9.
37. Álvarez-Pabón Yelitza. Leucodistrofia metacromática infantil tardía: Presentación de un caso. Arch Argent Pediatr. 2019;117(1).
38. Fumagalli DF, Fumagalli AI, Biffi A, Cesani M, Corea F, Sessa M, et al. Leucodistrofia metacromática: del diagnóstico a la terapia génica como perspectiva terapéutica. 1982;6.
39. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=ES&Expert=512. Orphanet: Leucodistrofia metacromática [Internet]. [cited 31 of December de 2018]. available in: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=ES&Expert=512
40. Ozgen H, Baron W, Hoekstra D, Kahya N. Oligodendroglial membrane dynamics in relation to myelin biogenesis. Cell Mol Life Sci. 2016;73:3291-310.
41. Vos JP, Lopes-Cardozo M, Gadella BM. Metabolic and functional aspects of sulfogalactolipids. Biochim Biophys Acta BBA - Lipids Lipid Metab. 1994;1211(2):125-49.
42. Sherman DL, Brophy PJ. Mechanisms of axon ensheathment and myelin growth. Nat Rev Neurosci. 2005;6(9):683-90.
43. Berntson Z, Hansson E, Ronnback L, Fredman P. Intracellular sulfatide expression in a subpopulation of astrocytes in primary cultures. J Neurosci Res. 1998;52(5):559-68.
44. Fullerton PM. Peripheral nerve conduction in metachromatic leucodystrophy (sulphatide lipidosis). J Neurol Neurosurg Psychiatry. 1964;27(2):100-5.
45. Popovich PG, Jakeman LB, McTigue DM. Glial Responses to Injury. En: Encyclopedia of Neuroscience [Internet]. Elsevier; 2009 [cited 26 July 2020]. p. 853-9. Available at: https://linkinghub.elsevier.com/retrieve/pii/B9780080450469000188
46. Brady ST, Witt AS, Kirkpatrick LL, de Waegh SM, Readhead C, Tu P-H, et al. Formation of Compact Myelin Is Required for Maturation of the Axonal Cytoskeleton. J Neurosci. 1999;19(17):7278-88.
47. Mikoshiba K, Okano H, Tamura TA, Ikenaka K. Structure and Function of Myelin Protein Genes. Annu Rev Neurosci. 1991;14(1):201-17.
48. Krämer E-M, Koch T, Niehaus A, Trotter J. Oligodendrocytes Direct Glycosyl Phosphatidylinositol-anchored Proteins to the Myelin Sheath in Glycosphingolipid-rich Complexes. J Biol Chem. 1997;272(14):8937-45.
49. Taylor CM, Coetzee T, Pfeiffer SE. Detergent‐insoluble glycosphingolipid/cholesterol microdomains of the myelin membrane. J Neurochem. 2002;81(5):993-1004.
50. Wong YC, Kim S, Peng W, Krainc D. Regulation and Function of Mitochondria–Lysosome Membrane Contact Sites in Cellular Homeostasis. Trends Cell Biol. 2019;29(6):500-13.
51. Plotegher N, Duchen MR. Crosstalk between Lysosomes and Mitochondria in Parkinson's Disease. Front Cell Dev Biol. 2017 [cited 11 April 2020];5. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5732996/
52. Deus CM, Yambire KF, Oliveira PJ, Raimundo N. Mitochondria–Lysosome Crosstalk: From Physiology to Neurodegeneration. Trends Mol Med. enero de 2020;26(1):71-88.
53. Platt FM, d’Azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nat Rev Dis Primer. 2018;4(1):27.
54. Biffi A, Lucchini G, Rovelli A, Sessa M. Metachromatic leukodystrophy: an overview of current and prospective treatments. Bone Marrow Transpl. 2008;42(2):275.
55. Lagranha VL, Baldo G, de Carvalho TG, Burin M, Saraiva-Pereira ML, Matte U, et al. In vitro correction of ARSA deficiency in human skin fibroblasts from metachromatic leukodystrophy patients after treatment with microencapsulated recombinant cells. Metab Brain Dis. 2008;23(4):469-84.
56. Eichler FS, Cox TM, Crombez E, Dali CÍ, Kohlschütter A. Metachromatic Leukodystrophy: An Assessment of Disease Burden. J Child Neurol. 2016;31(13):1457-63.
57. Bockenhoff A, Cramer S, Wolte P, Knieling S, Wohlenberg C, Gieselmann V, et al. Comparison of Five Peptide Vectors for Improved Brain Delivery of the Lysosomal Enzyme Arylsulfatase A. J Neurosci. 2014;34(9):3122-9.
58. Desnick RJ, Schuchman EH. Enzyme Replacement Therapy for Lysosomal Diseases: Lessons from 20 Years of Experience and Remaining Challenges. Annu Rev Genomics Hum Genet. 2012;13(1):307-35.
59. Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Benefits Metachromatic Leukodystrophy. Science. 2013;341(6148). Available at: http://science.sciencemag.org/content/341/6148/1233158.abstract
60. Kehrer C, Blumenstock G, Gieselmann V, Krägeloh-Mann I, GERMAN LEUKONET. The natural course of gross motor deterioration in metachromatic leukodystrophy. Dev Med Child Neurol. 2011;53(9):850-5.
61. Efficacy METAZYM for the Treatment Metachromatic Leukodystrophy Treated With Hematopoietic Stem Cell Transplantation - No Study Results Posted - ClinicalTrials.gov [Internet]. [cited 10 April 2020]. Available in: https://clinicaltrials.gov/ct2/show/results/NCT01303146
62. Shire. A Single Center, Open-label, Non-randomized, Uncontrolled, Multiple-dose, Dose Escalation Study of the Safety, Pharmacokinetics and Efficacy of Metazym for the Treatment of Patients With Late Infantile Metachromatic Leukodystrophy (MLD) [Internet]. clinicaltrials.gov; 2018 dic [citado 24 de julio de 2020]. Report No.: results/NCT00418561. Disponible en: https://clinicaltrials.gov/ct2/show/results/NCT00418561
63. Patil SA, Maegawa GH. Developing therapeutic approaches for metachromatic leukodystrophy. Drug Des Devel Ther. 2013;7:729-45.
64. Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol. 2009;10:623.
65. Intracerebral Gene Therapy for Children With Early Onset Forms of Metachromatic Leukodystrophy - Full Text View - ClinicalTrials.gov [Internet]. [cited 27 January 2019]. Available at: https://clinicaltrials.gov/ct2/show/NCT01801709
66. Asheuer M, Pflumio F, Benhamida S, Dubart-Kupperschmitt A, Fouquet F, Imai Y, et al. Human CD34+ cells differentiate into microglia and express recombinant therapeutic protein. Proc Natl Acad Sci U S A. 2004;101(10):3557-62.
67. Stem Cell Transplant for Inborn Errors of Metabolism - Full Text View - ClinicalTrials.gov [Internet]. [cited 27 January 2019]. Available at: https://clinicaltrials.gov/ct2/show/NCT00176904
68. Masonic Cancer Center, University of Minnesota. Treatment of Lysosomal and Peroxisomal Inborn Errors of Metabolism by Bone Marrow Transplantation [Internet]. clinicaltrials.gov; 2017 dic [cited 24 July 2020]. Report No.: study/NCT00176904. Available at: https://clinicaltrials.gov/ct2/show/study/NCT00176904
69. Rauschka H, Colsch B, Baumann N, Wevers R, Schmidbauer M, Krammer M, et al. Late-onset metachromatic leukodystrophy: Genotype strongly influences phenotype. Neurology. 2006;67(5):859-63.
70. Golebiowski D, Bradbury AM, Kwon C-S, van der Bom IMJ, Stoica L, Johnson AK, et al. AAV Gene Therapy Strategies for Lysosomal Storage Disorders with Central Nervous System Involvement. En: Bo X, Verhaagen J, editores. Gene Delivery and Therapy for Neurological Disorders [Internet]. New York, NY: Springer New York; 2015 [cited 27 January 2019]. p. 265-95. (Neuromethods). Available at: https://doi.org/10.1007/978-1-4939-2306-9_11
71. Rosenberg JB, Sondhi D, Rubin DG, Monette S, Chen A, Cram S, et al. Comparative Efficacy and Safety of Multiple Routes of Direct CNS Administration of Adeno-Associated Virus Gene Transfer Vector Serotype rh.10 Expressing the Human Arylsulfatase A cDNA to Nonhuman Primates. Hum Gene Ther Clin Dev. 1 de septiembre de 2014;25(3):164-77.
72. Rosenberg JB, Kaminsky SM, Aubourg P, Crystal RG, Sondhi D. Gene Therapy for Metachromatic Leukodystrophy. J Neurosci Res. 2016;94(11):1169-79.
73. Piguet F, Sondhi D, Piraud M, Fouquet F, Hackett NR, Ahouansou O, et al. Correction of Brain Oligodendrocytes by AAVrh.10 Intracerebral Gene Therapy in Metachromatic Leukodystrophy Mice. Hum Gene Ther. 2012;23(8):903-14.
74. Sondhi D, Hackett NR, Peterson DA, Stratton J, Baad M, Travis KM, et al. Enhanced Survival of the LINCL Mouse Following CLN2 Gene Transfer Using the rh.10 Rhesus Macaque-derived Adeno-associated Virus Vector. Mol Ther. 2007;15(3):481-91.
75. Sondhi D, Johnson L, Purpura K, Monette S, Souweidane MM, Kaplitt MG, et al. Long-Term Expression and Safety of Administration of AAVrh.10hCLN2 to the Brain of Rats and Nonhuman Primates for the Treatment of Late Infantile Neuronal Ceroid Lipofuscinosis. Hum Gene Ther Methods. 2012;23(5):324-35.
76. Ormond KE, Mortlock DP, Scholes DT, Bombard Y, Brody LC, Faucett WA, et al. Human Germline Genome Editing. Am J Hum Genet. 2017;101(2):167-76.
77. Thurtle‐Schmidt DM, Lo T. Molecular biology at the cutting edge: A review on CRISPR/CAS9 gene editing for undergraduates. Biochem Mol Biol Educ. 2018;46(2):195-205.
78. Ashrafi MR, Amanat M, Garshasbi M, Kameli R, Nilipour Y, Heidari M, et al. An update on clinical, pathological, diagnostic, and therapeutic perspectives of childhood leukodystrophies. Expert Rev Neurother. 2020;20(1):65-84.
79. Manual de Pruebas Diagnosticas de Laboratorio Clinico. Alejandro Almarza; 130 p.
80. Winzeler AM, Mandemakers WJ, Sun MZ, Stafford M, Phillips CT, Barres BA. The Lipid Sulfatide Is a Novel Myelin-Associated Inhibitor of CNS Axon Outgrowth. J Neurosci. 2011;31(17):6481-92.
Received:15 arch 2021
Accepted: 10 July 2021
Nayibe Tatiana Sanchez-Alvarez 1.2.3; Paula Katherine Bautista-Niño 2; Juanita Trejos-Suárez 3, Norma Cecilia Serrano-Diaz2
1 Universidad del Valle, Faculty of Health,
Biomedical Sciences Doctorate Program,
2 Colombian Cardiovascular Foundation, Research Center.
Floridablanca, Santander, Colombia.
3Universidad de Santander, Faculty of Health Sciences, CliniUDES Research Group, Bucaramanga, Santander, Colombia.
Corresponding author: Nayibe Tatiana Sanchez Alvarez, Ph.D. Student in Biomedical Sciences, Universidad del Valle (UNIVALLE), Faculty of Health; Cardiovascular Foundation of Colombia, Cra. 5 # 6 - 33, Floridablanca, Santander, Colombia. 3183892918, [email protected]