2023.08.01.27
Files > Volume 8 > Vol 8 No 1 2023
Identificación de inhibidores de las enzimas RdRp y Mpro del virus SARS-CoV-2 mediante homología estructural
Identification of inhibitors of the RdRp and Mpro enzymes of SARS-CoV-2 virus by structural homology

Daysi Espín-Sánchez1* , María L. Ramos-Aristimbay1*
, María L. Ramos-Aristimbay1* , Andrés S. Sánchez-Vaca1*
, Andrés S. Sánchez-Vaca1* , Karen Jaramillo-Guapisaca1
, Karen Jaramillo-Guapisaca1 , Carolina Vizueta-Rubio1
, Carolina Vizueta-Rubio1 , Fernanda Chico-Terán3
, Fernanda Chico-Terán3 , Liliana Cerda-Mejía2*
, Liliana Cerda-Mejía2* , Mario D. García1, 3*
, Mario D. García1, 3* .
.
1Carrera de Ingeniería Bioquímica, Facultad de Ciencia e Ingeniería en Alimentos y Biotecnología Universidad Técnica de Ambato
2Carrera de Alimentos, Facultad de Ciencia e Ingeniería en Alimentos y Biotecnología Universidad Técnica de Ambato
3Carrera de Biotecnología, Facultad de Ciencia e Ingeniería en Alimentos y Biotecnología Universidad Técnica de Ambato
* Estos autores contribuyeron de igual forma.
* Autores de correspondencia Email:
*Daysi Espín-Sánchez: [email protected]
*María L. Ramos-Aristimbay: [email protected]
*Andrés S. Sánchez-Vaca: [email protected]
*Mario D. García: [email protected]
Available from: http://dx.doi.org/10.21931/RB/2023.08.01.27
ABSTRACT
El COVID-19 ha generado un enorme impacto en la salud pública mundial debido a las altas tasas de contagio y mortalidad asociadas al virus SARS-CoV-2 causante de la enfermedad. Hasta la fecha, la Organización Mundial de la Salud (OMS) ha aprobado el uso de 10 vacunas aparentemente seguras y eficaces. Sin embargo, todavía existen limitaciones importantes para su administración en países en vías de desarrollo y localidades remotas, y la preocupación por la aparición de variantes del virus que puedan evadir la inmunidad adquirida mediante la vacunación se mantiene latente. Además de la prevención de la infección, son necesarios agentes terapéuticos efectivos para tratar a los pacientes diagnosticados con COVID-19. Bajo este contexto, el presente estudio tuvo como objetivo realizar un cribado virtual basado en la estructura de las enzimas proteasa (Mpro) y ARN polimerasa ARN-dependiente (RdRp) del SARS-CoV-2. Para este propósito se ensayaron inhibidores de proteínas homólogas pertenecientes a diferentes virus. El alineamiento múltiple de secuencias de estas enzimas permitió reconocer la presencia de una alta conservación de estas enzimas entre especies, especialmente de las regiones que comprenden los sitios de unión a inhibidores. Por lo tanto, se deduce que es posible emplear un enfoque de redireccionamiento de los inhibidores que fueron diseñados para tratar otras enfermedades virales. Experimentos de acoplamiento molecular permitieron identificar que los inhibidores RTP (afinidad de unión = -7.3 kcal/mol) y V3D (afinidad de unión = -8.0 kcal/mol) son excelentes inhibidores de RdRp y Mpro, respectivamente. Estos resultados sugieren que dichas moléculas son virtualmente capaces de unirse e inhibir la actividad de RdRp y Mpro y por lo tanto constituyen potenciales fármacos para combatir el SARS-CoV-2.
Palabras clave: SARS-CoV-2, COVID-19, inhibidores, RdRp, Mpro.
ABSTRACT
COVID-19 has enormously impacted global public health due to the high infection and mortality rates associated with the SARS-CoV-2 virus-causing disease. The World Health Organization (WHO) approved 10 safe and effective vaccines. However, there are still significant limitations to their administration in developing countries and remote locations. Concerns remain about the emergence of virus variants that may evade immunity acquired through vaccination. In addition to preventing infection, effective therapeutic agents are needed to treat patients diagnosed with COVID-19. Under this context, the present study aimed to perform a structure-based virtual screening of the protease (Mpro) and RNA-dependent RNA polymerase (RdRp) enzymes of SARS-CoV-2. For this purpose, homologous protein inhibitors belonging to different viruses were tested. Multiple sequence alignment of these enzymes allowed us to recognize the high conservation of these enzymes between species, especially of the regions comprising the inhibitor binding sites. Therefore, it follows that it is possible to employ a redirection approach to inhibitors that were designed to treat other viral diseases. Molecular docking experiments identified that RTP inhibitors (binding affinity = -7.3 kcal/mol) and V3D (binding affinity = -8.0 kcal/mol) are excellent inhibitors of RdRp and Mpro, respectively. These results suggest that these molecules can virtually bind and inhibit the activity of RdRp and Mpro and thus constitute potential drugs to combat SARS-CoV-2.
Keywords: SARS-CoV-2, COVID-19, inhibitors, RdRp, Mpro.
INTRODUCCIÓN
La COVID-19, causada por el SARS-CoV-2, es una enfermedad viral emergente de alta preocupación por su elevado impacto sobre la salud pública y la economía global. En Marzo de 2020, la enfermedad fue declarada pandemia tras su rápida propagación alrededor de todo el mundo, llevando a una emergencia sanitaria que continúa vigente1. Hasta el momento de redacción de este artículo, según cifras oficiales, a nivel global se han confirmado más 437 millones de casos de COVID-19, incluyendo 5,96 millones muertes2, estos datos indican que su tasa de mortalidad (CFR) asociada es de 1,36%.
El SARS-CoV-2 taxonómicamente pertenece al reino Riboviria, orden Nidovirales, familia Coronaviridae y género Betacoronaviridae, clasificación correspondiente a virus que poseen ARN de hebra sencilla positiva, envoltura de membrana y que tienen la capacidad de infectar humanos3. Análisis filogenéticos del genoma completo del SARS-CoV-2 evidencian su cercanía con coronavirus de murciélago y con el SARS-CoV, presentando entre sí más del 96% de identidad de secuencia4 5. Así mismo, se ha revelado que su genoma tiene una longitud aproximada de 30 kb, el cual consiste en dos regiones no traducidas (UTR) y 14 marcos de lectura abierta (ORFs). El primer ORF (Orf 1/ab) abarca más de la mitad del genoma y codifica 16 proteínas no estructurales (NSP) del complejo de replicación-transcripción, mientras que los 13 ORFs restantes codifican cuatro proteínas estructurales y al menos 6 proteínas accesorias5 6 7 8.
Los productos del genoma pueden ser blancos moleculares de agentes antivirales, puesto que se ha demostrado que cumplen funciones críticas en el ciclo de vida de los coronavirus6. Las dianas más prometedoras en el SARS-CoV-2 parecen ser las proteínas proteasa principal (Mpro), proteasa de tipo papaína (PLpro) y la ARN polimerasa ARN dependiente (RdRp), ya que comparten el mayor porcentaje de homología con otros coronavirus9. También se ha considerado a la proteína espiga (S) como una diana terapéutica, por ser la responsable de la primera interacción del virus con las células del huésped. Sin embargo, esta proteína es altamente susceptible a mutaciones10.
Tras el ingreso del virus a las células, el genoma viral es transcrito a poliproteínas del complejo de replicación/transcripción (RTC), que son procesadas mediante las proteasas Mpro y PLpro para generar proteínas efectoras funcionales11, siendo Mpro la enzima predominante en la transformación proteolítica12. Un componente clave del RTC es la enzima RdRp, encargada de sintetizar ARN genómico para el ensamblaje de nuevas partículas del virus13 14. Tanto Mpro como RdRp presentan más del 95% de similitud de secuencia y estructura con las enzimas del SARS-CoV14 y se sabe que no poseen homólogos cercanamente relacionados en humanos15 16 por lo que pueden ser importantes dianas para el desarrollo de inhibidores con alta especificidad17.
Debido al enorme impacto que ha tenido el COVID-19 en la salud pública mundial, el desarrollo y producción de medicamentos específicos y vacunas es urgente18. En la actualidad existen 10 vacunas seguras y eficaces, cuyo uso fue aprobado por la OMS19. No obstante, a pesar de la creciente disponibilidad de vacunas contra la COVID-19, aún existen limitaciones importantes para su administración, sobre todo en zonas del planeta con escasos recursos económicos20 21, y su efectividad contra las nuevas variantes del virus es variable.
Además de la disminución de la transmisión comunitaria del virus y prevención de la infección, son necesarios también agentes terapéuticos efectivos para tratar a los pacientes que han desarrollado cuadros severos de la enfermedad22. Debido a la similitud existente entre las enzimas del SARS-CoV-2 con virus como el SARS-CoV e incluso MERS-CoV, los fármacos utilizados para tratar estas y otras infecciones del mismo tipo se encuentran disponibles y pueden ser útiles en la lucha contra el COVID-1918. Bajo este contexto, los esfuerzos actuales están enfocados en el redireccionamiento de fármacos con eficacia probada en contra de virus homólogos al SARS-CoV-223, lo cual ha conducido a ensayar medicamentos que inhiben a RdRp, tales como Faripiravir y Remdesivir, éste último es un análogo de nucleósido que a mediados del 2020 obtuvo la aprobación de uso de emergencia por la US FDA en pacientes con COVID-19 severo24. Así mismo, otros ejemplos de fármacos son Carmofur y N3, que han demostrado capacidad de inhibición selectiva de Mpro de múltiples coronavirus, lo que los hacen efectivos antivirales con potencial actividad sobre el SARS-CoV-225.
El cribado virtual de librerías de compuestos puede ser una estrategia interesante para descubrir antivirales que permitan paliar los efectos adversos de la enfermedad26. De este modo, se puede acortar la lista de fármacos con aquellos que parecen unirse con mayor afinidad a las enzimas virales27 y de los cuales se conocen características como el perfil de seguridad, efectos secundarios y posología22. Herramientas de acoplamiento molecular (molecular docking) permiten efectuar el cribado virtual de compuestos previo a su estudio in-vitro e in-vivo, haciendo posible la predicción de la orientación del fármaco en un receptor particular y su energía de unión. Mediante esta perspectiva, se pueden evaluar cientos de moléculas a un bajo costo y en relativamente poco tiempo28 29. En el presente estudio se realizó un cribado virtual de inhibidores basado en la estructura de enzimas Mpro y RdRp pertenecientes a virus estructuralmente homólogos al SARS-CoV-2. Este enfoque tuvo como objetivo identificar fármacos con potencial para ser redireccionados hacia el tratamiento de pacientes diagnosticados con COVID-19, o a su vez, para ser empleados en el diseño racional de nuevos inhibidores específicos de Mpro y RdRp del SARS-CoV-2.
MATERIALES Y MÉTODOS
Análisis de homología estructural
A partir de las estructuras 3D de las enzimas RdRp (PDB ID: 6m71) y Mpro (PDB ID: 6yb7) del SARS-CoV-2 se identificó, mediante el uso del servidor PDBeFold30, enzimas virales que comparten similitud estructural según el parámetro SSE (matched Secondary Structure Elements). En el caso de la polimerasa solo se seleccionó la cadena principal de la proteína (subunidad catalítica nsp12). De los resultados se tomó únicamente las estructuras en complejo con inhibidores.
Construcción de la librería de inhibidores de enzimas Mpro y RdRp
De los datos arrojados por PDBeFold se identificaron inhibidores acoplados a proteínas RdRp y Mpro homólogas a aquellas del SARS-CoV-2. La estructura química de cada inhibidor fue obtenida a partir del complejo enzima-ligando depositado en el PDB31 y sus coordenadas tridimensionales fueron exportadas mediante PyMOL32.
Identificación de los sitios de unión de inhibidores
Los sitios de unión de los diferentes inhibidores y sus interacciones con la proteína fueron determinados mediante su superposición con RdRp o Mpro del SARS-CoV-2 en WinCoot33 y PyMOL32. Para el análisis se consideró aquellas interacciones no covalentes en el rango de 2.2 a 4.2 Å.
Alineamiento múltiple de secuencias
Tras la identificación de proteínas homólogas, se realizó un alineamiento múltiple de sus secuencias mediante T-Coffee34. El alineamiento fue posteriormente coloreado y analizado mediante BoxShade de ExPASy35. En el alineamiento se establecieron los principales aminoácidos involucrados en el acoplamiento de ligandos de cada enzima.
Preparación de proteínas y ligandos
La preparación de los receptores RdRp (PDB ID: 7bv2) y Mpro (PDB ID: 7bqy), así como de la librería de ligandos (120 compuestos) se realizó utilizando AutoDock Tools versión 1.5.6 rc236, obteniendo de este modo las estructuras 3D en formato pdbqt.
Molecular docking
Los ensayos de acoplamiento molecular no covalente rígido se llevaron a cabo usando AutoDock Vina en PyRx37, 38. Se utilizó un grid apropiado para cada uno de los ligandos y se usó un valor de exhaustividad igual a 8. Las interacciones enzima-ligando fueron analizadas con Discovery Studio Visualizer39 y el modo de unión de los compuestos se determinó mediante PyMOL32. Las ilustraciones fueron elaboradas mediante PyMOL y Discovery Studio Visualizer.
RESULTADOS Y DISCUSIÓN
Análisis de homología estructural
El análisis de homología estructural permitió identificar varias enzimas polimerasas y proteasas que comparten elementos estructurales con la subunidad nsp12 de RdRp y con Mpro del SARS-CoV-2. El valor más alto de %SSE observado para la RdRp corresponde a la polimerasa del virus Zika (ZV), enzima que comparte el 49% de similitud de estructura secundaria con su homóloga del SARS-CoV-2. Por otra parte, las polimerasas del virus de la fiebre aftosa humana (HFMD), Coxsackievirus (CV), Norovirus murino (NVM) y virus Norwalk (NV) presentan un 47% de similitud estructural. Finalmente, se observó un 45% de homología estructural con las RdRp de los virus del Dengue (DV) y de la Hepatitis C (HCV). La baja conservación de los elementos de estructura secundaria entre la RdRp del SARS-CoV-2 y enzimas homólogas se puede deber a la gran diferencia de tamaño que existe entre estas polimerasas. Se conoce que las características principales y la longitud (500 residuos) de las RdRp virales es altamente conservada pese a su baja identidad de secuencia40. Sin embargo, la subunidad nsp12 de la RdRp del SARS-CoV-2, con 932 residuos, es significativamente más larga que sus homólogas estructurales que cuentan con secuencias de ~500 aminoácidos. Aquí, hemos observado que la posición y la arquitectura general de los dominios y del sitio activo son altamente similares en las diferentes RdRps, lo que sugiere que los inhibidores de RdRps se unen a estas enzimas de forma similar y que la diferencia en la longitud de los polipéptidos no afecta el modo unión de los inhibidores.
Para Mpro, el análisis estructural determinó que existen enzimas virales de organismos homólogos, como Mpro del SARS-CoV, que comparten hasta un 100% de similitud estructural. De igual forma, el Coronavirus de murciélago HKU4 (HKU4-CoV) comparte un 95% de similitud, mientras que los organismos Coronavirus humano NL63 (HCoV-NL63), MERS-CoV y Virus de la diarrea epidémica porcina (PEDV) muestran hasta un 91% de identidad de estructura secundaria. Por último, los coronavirus de la peritonitis infecciosa felina (FIPV) y de la gastroenteritis transmisible porcina (TGEV) exhiben un 86% y 82% de semejanza, respectivamente. A pesar de la evidente conservación de la enzima, un estudio detallado sobre Mpro, basado en análisis de secuencia y estructura, ha revelado que ciertas variantes del virus SARS-CoV-2 presentan unas pocas mutaciones puntuales. Dicho estudio además sugiere que tales cambios probablemente surgieron como respuesta a la evolución adaptativa41. En conclusión, el análisis de homología estructural sugiere que los elementos de estructura secundaria son mucho más conservados en Mpro que en RdRp. Sin embargo, las características de sus sitios de unión a inhibidores son altamente conservados en ambas enzimas lo cual sustenta la hipótesis de que es posible redireccionar moléculas diseñadas para inhibir la proteasa o polimerasa de virus homólogos al SARS-CoV-2.
Identificación de inhibidores de enzimas RdRp y Mpro en virus homólogos al SARS-CoV-2
El análisis de homología estructural también permitió identificar un total de 78 moléculas que actúan como inhibidores de las enzimas RdRp de ZV, HFMD, CV, NVM, NV, HCV y DV, donde la mayoría de los inhibidores descritos pertenecen a la polimerasa del HCV (ns5b HCV). Para la enzima Mpro se identificaron 41 inhibidores de los virus SARS-CoV, HKU4-CoV, HCoV-NL63, MERS-CoV, PEDV, FIPV y TGEV. La mayor parte de estos inhibidores pertenecen al SARS-CoV. El listado de ligandos obtenidos del análisis de homología estructural se muestra en la Tabla Suplementaria 1.
En el caso de la RdRp el análisis individual ha mostrado que ciertos inhibidores se unen covalentemente a diferentes aminoácidos o a la plantilla de ARN durante la síntesis. No obstante, la gran mayoría lo hace mediante enlaces no covalentes de tipo puente de hidrógeno y fuerzas de Van der Waals. En Mpro, todos los inhibidores identificados forman contactos con el residuo Cys145, el cual se encuentra altamente conservado entre diferentes especies. La unión se da tanto de forma no covalente y covalente, predominando esta última. Los inhibidores reportados para las dos enzimas muestran amplios rangos de afinidad de unión (IC50, Ki o Kd) en el rango nM a mM (Tabla Suplementaria 1). En cuanto a su estructura química, entre los inhibidores destacan compuestos de diferente índole, como ácidos carboxílicos, ácidos sulfónicos, ácidos benzoicos, cetonas, amidas, aminas y nitrilos. Se identificó varias estructuras químicas relevantes, como las sulfonamidas y carboxamidas. Se conoce que las sulfonamidas constituyen el esqueleto para el diseño de un amplio rango de agentes terapéuticos. A partir de las sulfonamidas se han elaborado antibacteriales, anticancerígenos, anticonvulsionantes, antidiabéticos, diuréticos, antileucémicos, antiinflamatorios y antivirales. De igual forma las carboxamidas son importantes farmacóforos que se encuentran en moléculas de medicamentos usados para regular los niveles de colesterol, hipertensión, enfermedades del corazón y el VIH, entre otras enfermedades42.
Un gran número de los inhibidores específicos identificados para RdRps y que actúan sobre el sitio activo son análogos de nucleósidos. La enzima RdRp del SARS-CoV-2, al igual que otras polimerasas, posee baja fidelidad, por lo que su propensión a errores durante el proceso de replicación incrementa la posibilidad de aceptación de análogos de nucleótidos como sustratos. Por esta razón, los nucleótidos y nucleósidos modificados son importantes agentes antivirales ya que inhiben la enzima al competir con los ribonucleótidos naturales15.
Para Mpro, dada su función proteolítica, los primeros inhibidores reportados con capacidad de unirse covalentemente al sitio activo fueron peptidomiméticos, que son compuestos análogos del sustrato que asemejan una estructura proteica y que además poseen grupos reactivos para atacar al residuo catalítico Cys14517. Como era de esperarse, el presente estudio reporta también la aparición de este tipo de moléculas (Tabla Suplementaria 1). Además, aquí identificamos que los carbamatos también constituyen un grupo representativo de inhibidores en diferentes enzimas Mpro. Estos compuestos, en particular los orgánicos, juegan un rol importante en el descubrimiento de fármacos y constituyen elementos estructurales de diversos agentes terapéuticos de funcionalidad probada como ritonavir, amprenavir, atazanavir y darunavir43. Algunas de las características que hacen a los carbamatos atractivos para el diseño de fármacos son su elevada estabilidad, permeabilidad a las membranas celulares y su habilidad para regular interacciones con las enzimas objetivo44.
Definición de los sitios de unión a inhibidores
La enzima RdRp del SARS-CoV-2, a pesar de sus diferencias estructurales con otras del mismo tipo, mantiene la configuración tridimensional típica de polimerasas denominada de “mano derecha”, pues comprende los dominios conocidos como Palma (Palm), Dedos (Finger) y Pulgar (Thumb) (Figura 1B). Interesantemente, se ha reportado que varias RdRps pueden ser inhibidas por moléculas que se unen al sitio activo y a múltiples bolsillos alostéricos. El presente estudio permitió determinar que se dispone de una gran cantidad de información estructural de la enzima ns5b HCV en complejo con diferentes inhibidores. ns5b HCV posee una arquitectura tridimensional característica, donde el sitio activo se ubica en el dominio de la Palma, que implica la cavidad central de la enzima, y donde también se ubican dos sitios adicionales de unión a inhibidores (Palm I site y Palm II site). De igual manera en el dominio del Pulgar se han identificado al menos tres sitios alostéricos (Thumb I site, Thumb II site y Thumb III site) que permiten acoplar ligandos28, 45(Figura 1A). Respondiendo al grado de conservación estructural de RdRps, en organismos como ZV, NV y NVM también se han reconocido inhibidores alostéricos que se unen en bolsillos cercanos al sitio activo. En contraste, los inhibidores que se han reportado para la nsp12 RdRp actúan únicamente tras su unión en el sitio activo ubicado en dominio de la Palma46 (Figura 1B).
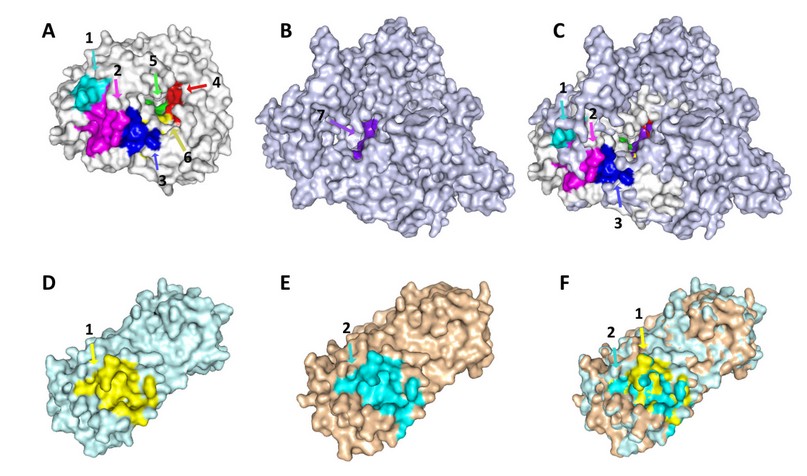
Figura 1. Comparación estructural de sitios de unión a inhibidores en enzimas del SARS-CoV-2. A) Se muestran los 6 bolsillos de unión de la RdRp ns5b HCV: en color cyan el sitio alostérico Thumb I (1), en color magenta el sitio alostérico Thumb II (2), en color azul el sitio alostérico Thumb III (3), en color rojo el sitio activo de la enzima (4), en color verde el sitio alostérico Palm I (5) y en color amarillo el sitio alostérico Palm II (6). B) Se muestra el único bolsillo de unión de la RdRp nsp12 del SARS-CoV: en color morado el sitio activo (7). C) La superposición entre ns5b HCV y nsp12 del SARS-CoV-2. D) Se muestra el sitio activo de la enzima Mpro del SARS-CoV en color amarillo (1). E) Se muestra el sitio activo de la enzima Mpro del SARS-CoV-2 en color cyan (2). F) Superposición entre Mpro del SARS-Co-V y SARS-CoV-2.
La superposición estructural de la RdRp del SARS-CoV-2 con su homóloga de referencia (ns5b HCV) (Figura 1C), muestra que en ambas enzimas existe correspondencia de sitios de unión respecto a todos aquellos bolsillos ubicados en la cavidad central de la Palma (Palm I y Palm II), así como de un sitio alostérico en el dominio del Pulgar (Thumb III), pues, aunque se aprecian diferencias en su conformación, estos son visibles en la estructura. Por el contrario, los dos bolsillos alostéricos restantes (Thumb I y Thumb II) prácticamente son inexistentes en la polimerasa del SARS-CoV-2, donde los aminoácidos de la cadena peptídica sobresalen de la estructura impidiendo la formación de cavidades que podrían acoplar inhibidores.
De esta forma, con base en la semejanza estructural con sus enzimas homólogas, en el presente estudio se ensayó el acoplamiento molecular de los ligandos en los bolsillos sitio activo, Palm I site, Palm II site y Thumb III site de RdRp del SARS-CoV-2, asumiendo la posible existencia de estos tres últimos sitios alostéricos. La superposición de ns5b y nsp12 (Figura 1C), permitió determinar de manera visual los aminoácidos que conforman los sitios alostéricos teóricos de la RdRp del SARS-CoV-2, mismos que se indican en la Tabla 1.
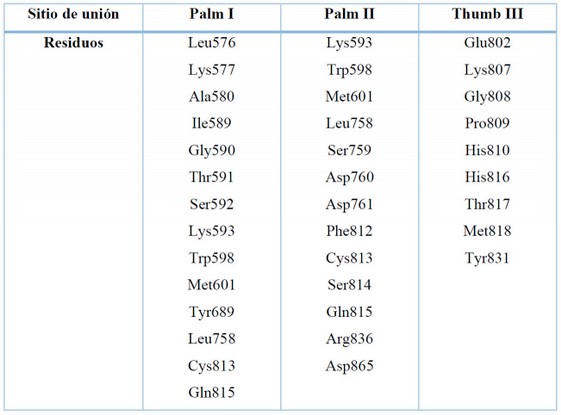
Tabla 1. Aminoácidos que conforman los bolsillos alostéricos teóricos de la RdRp del SARS-CoV-2.
Por su parte, Mpro es una cisteína proteasa cuyo sitio activo está compuesto por cuatro sub-sitios denominados S1´, S1, S2 y S4. Esta enzima consta de tres dominios (I-III) y presenta una diada catalítica Cys-His no canónica localizada en la hendidura que se forma entre los dos primeros dominios47. Un estudio de cribado de pequeñas moléculas ha permitido caracterizar compuestos inhibidores alostéricos de Mpro que pueden unirse en un bolsillo distante del sitio activo y que son capaces de modular la actividad enzimática48. No obstante, debido a la gran conservación estructural de la enzima, los inhibidores que se unen al sitio activo comparten entre sí numerosas características47, lo cual es enormemente favorable para el redireccionamiento de fármacos. Por lo tanto, en la presente investigación únicamente se han considerado inhibidores que se acoplan en el sitio activo clásico para los ensayos de docking (Tabla Suplementaria 1). Como se mencionó anteriormente, el análisis de homología estructural mostró que Mpro del SARS-CoV (Figura 1D) posee una configuración tridimensional virtualmente idéntica con respecto a Mpro del SARS-CoV-2 (Figura 1E). La superposición de ambas proteasas demostró que sus sitios activos poseen una arquitectura altamente similar (Figura 1F), lo cual sugiere que los inhibidores que tienen como blanco este bolsillo, podrían acoplarse de igual forma a ambas enzimas.
Alineamiento múltiple de secuencias
Para la polimerasa del SARS-CoV-2, el primer inhibidor para el cual se descubrió experimentalmente la estructura del complejo enzima-ligando fue Remdesivir Monofosfato (RMP). Mediante el análisis de sus interacciones con la enzima se determinó que el sitio activo se ubica en los motivos A-G del dominio de la Palma10, y está conformado principalmente por 14 aminoácidos. El alineamiento múltiple de enzimas RdRp (Figura Suplementaria 1) muestra que la mayoría de los residuos involucrados en el acoplamiento del ligando, tales como Arg555, Asp618, Asp623, Ser682, Thr687, Asn691, Asp760 y Asp761, están altamente conservados entre los diferentes organismos analizados. Por otra parte, Val557 está sólo medianamente conservada, mientras que Arg553, Lys545, Thr680, Ala688 y Ser759 no muestran ningún grado de conservación. Esto señala que a pesar de las diferencias de tamaño de las secuencias de las diferentes RdRps homólogas a la polimerasa del SARS-CoV-2, los principales aminoácidos que intervienen en la unión al sustrato e inhibidores competitivos se mantienen entre especies. Por lo tanto, esto sugiere que los inhibidores competitivos de las polimerasas de ZV, HFMD, CV, NVM, NV, HCV y DV pueden ser alternativas viables para inhibir la replicación del SARS-CoV-2.
La ns5b HCV fue utilizada como referencia debido a la amplia información estructural disponible y cantidad de inhibidores descritos, por lo que los aminoácidos que conforman sus diferentes bolsillos de unión a ligandos también fueron comparados e identificados en nuestro análisis (Figura Suplementaria 1). Esto permitió determinar que 8 (Arg555, Asp618, Asp623, Ser682, Thr687, Asn691, Asp760 y Asp761) de los 13 aminoácidos que conforman el sitio activo de la ns5b están conservados en la nsp12. Estos residuos forman parte del centro catalítico.
Para los 5 bolsillos alostéricos adicionales de la ns5b; sin embargo, no se obtuvo una adecuada correspondencia entre aminoácidos, lo que significa que al menos en términos de secuencia estos no se encuentran conservados. Únicamente para los sitios alostéricos Palm I, Palm II y Thumb III de ns5b se pudo observar la presencia de bolsillos similares en nsp12, considerados aquí como sitios alostéricos teóricos, y por lo tanto incluidos en los estudios de acoplamiento molecular.
En cuanto a la proteasa, tomando como referencia a la estructura de la Mpro del virus SARS-CoV-2 unida al inhibidor N3, en el presente estudio se identificaron un total de 21 aminoácidos que participan en el acoplamiento del sustrato al sitio activo. Estos aminoácidos ayudan a estabilizar la unión y a bloquear el compuesto al interior de la cavidad mediante múltiples enlaces no covalentes25. Como se indica en la Figura Suplementaria 2, se pudo determinar que todos los organismos incluidos en el alineamiento múltiple de secuencias muestran conservación del sitio de unión de N3. Específicamente, los residuos de Thr26, Leu27, His41, Phe140, Asn142, Gly143, Ser144, Cys145, His163, Met165, Glu166, Pro168, His172, Asp187, Arg188, Thr190 y Glu192 son extremadamente conservados. En este grupo resaltan los aminoácidos catalíticos Cys145 e His41, que por su función central se mantienen conservados en todas las especies. En contraste, solamente 4 residuos, Met49, Leu141, His164 y Gln189, muestran un bajo grado de conservación. De esta forma, el alineamiento múltiple de secuencias demostró que el sitio activo y el bolsillo de unión de N3 es significativamente conservado entre enzimas Mpro de diferentes organismos. Esto sumado a los elevados porcentajes de similitud arrojados por el análisis de homología estructural, permiten deducir que los inhibidores de las enzimas proteasas de los virus SARS-CoV, HKU4-CoV, HCoV-NL63, MERS-CoV, PEDV, FIPV y TGEV, son excelentes candidatos para el tratamiento del COVID-19, o bien para el diseño racional de nuevos compuestos.
Acoplamiento molecular
El cribado virtual mediante acoplamiento molecular se basó en la determinación de la energía libre de unión (Binding affinity) de los complejos enzima-ligando38. Utilizando RdRp como receptor se ensayó el acoplamiento molecular de 78 inhibidores, los cuales mostraron valores de afinidad de unión en el rango de -2 a -7.4 kcal/mol (Tabla suplementaria 2). Los tres mejores resultados fueron XNI (-7.4 kcal/mol), RTP (Ribavirin Trifosfato) (-7.3) y JT1 (JTK-853) (-7.1 kcal/mol) (Tabla 2). La estructura química de estos compuestos se detalla en la Figura 2.
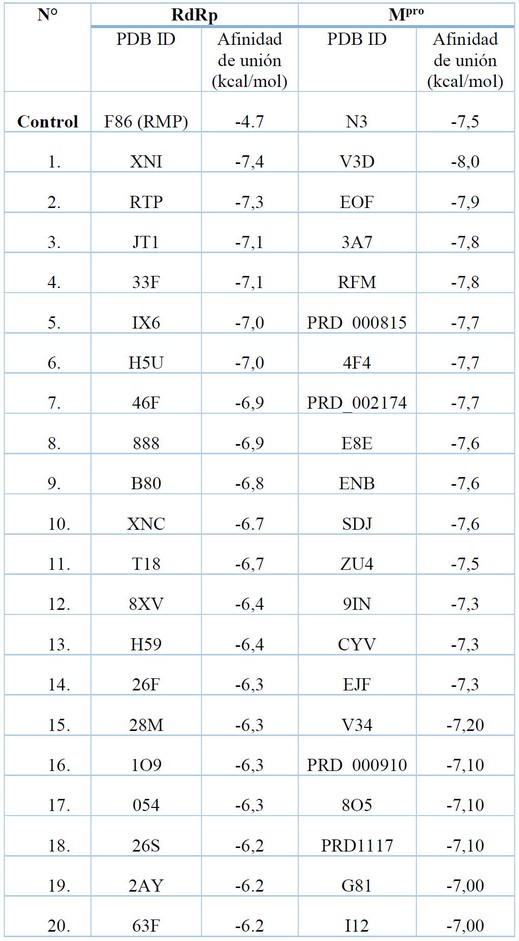
Tabla 2. Bestranking de resultados del acoplamiento molecular de ligandos obtenidos mediante homología estructural en RdRp y Mpro del virus SARS-CoV-2.
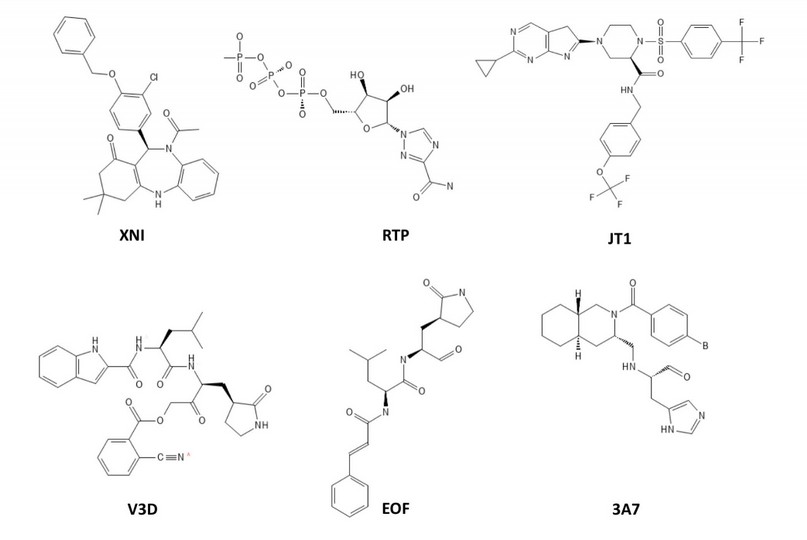
Figura 2. Estructura química de los mejores resultados del acoplamiento molecular en enzimas RdRp y Mpro del SARS-CoV-2. XNI, RTP y JT1 son inhibidores de la RdRp; V3D, EOF y 3A7 son inhibidores de Mpro.
El análisis de las coordenadas tridimensionales generadas mediante el acoplamiento molecular permitió evidenciar que XNI y JT1 se acoplan al sitio de la Palma I, mientras que RTP y el control, RMP, se unen al sitio activo de la enzima. Cabe recalcar que los inhibidores de la nsp12 reportados hasta la fecha se unen en al sitio activo y todavía no existe evidencia de inhibidores capaces de unirse a sitios alostéricos adicionales. En este sentido, dada la homología de estructura primaria y secundaria que guardan los sitios activos de las polimerasas del HFMD y SARS-CoV-2 (Figura 4B), RTP puede acoplarse a la nsp12 de forma similar al control, RMP. De hecho, RPT muestra un valor de afinidad superior a aquel observado para el control (-4.7 kcal/mol). Por lo tanto, RTP parece ser el resultado más relevante, pues su modo de unión se asemeja a los compuestos con actividad inhibitoria y antiviral in-vitro e in-vivo, tales como RMP. RTP es un análogo de guanosina, que difiere de ésta porque la D-ribosa se encuentra unida a un anillo de 1,2,4-triazol-carboxamida en lugar de un anillo de purina, y en su forma activa (trifosfatada), interfiere con la replicación viral49, lo cual explica su afinidad por el sitio activo de la nsp12.
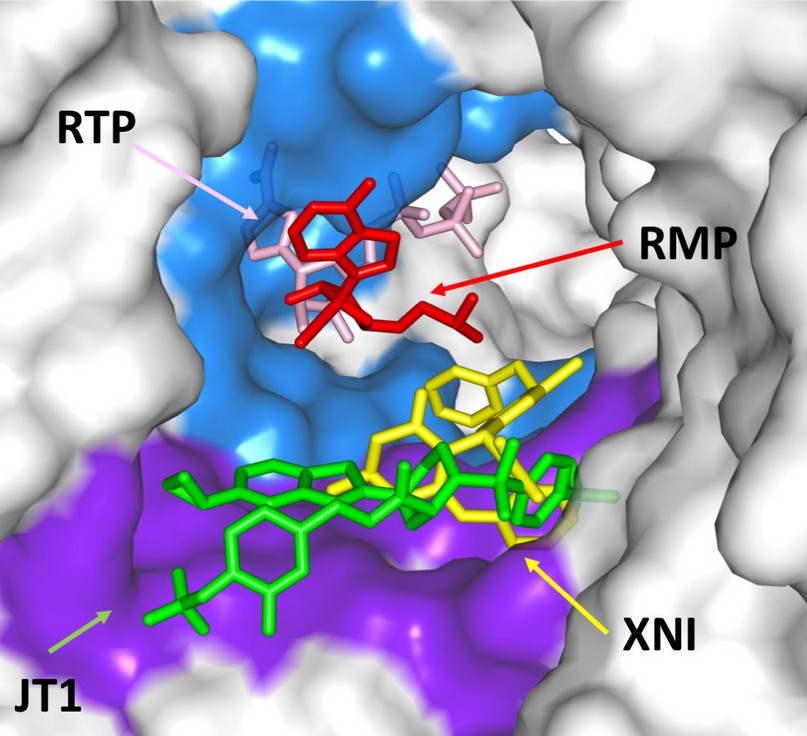
Figura 3. Modo de unión a inhibidores de la enzima RdRp del SARS-CoV-2 según el acoplamiento molecular. Se muestran en palillos los inhibidores XNI, RTP, JT1 y la molécula control Remdesivir Monofosfato (RMP), y en representación de superficie la enzima, en color azul el sitio activo de la enzima y en color morado el sitio alostérico teórico Palm I. Por otra parte, XNI (Ki = 1.7 μM) y JT1 (IC50 = 17.8 nM) son inhibidores reversibles que se unen en el sitio alostérico Palm I de la ns5b HCV (Figura 3). Si bien el alineamiento de secuencias demostró que los aminoácidos que componen este bolsillo de unión no se conservan en la nsp12, la superposición del sitio de la Palma I de ns5b junto con la estructura de la nsp12 muestra un bolsillo similar en la polimerasa del SARS-CoV-2 (Figura 4A).
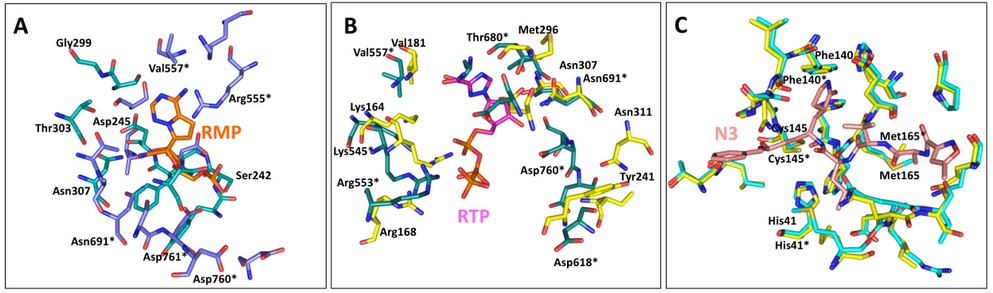
Figura 4. Comparación del sitio de unión a inhibidores entre enzimas del SARS-CoV-2 y sus homólogas de referencia. A) Superposición del sitio alostérico Palm I entre RdRp del HCV en palillos de color verde y del SARS-CoV-2 en palillos de color morado; se indica en palillos de color naranja el inhibidor RMP. B) Superposición del sitio activo de RdRp de HFMD en palillos de color amarillo y del SARS-CoV-2 en palillos de color verde; se indica en palillos de color magenta el inhibidor RTP. C) Superposición del sitio activo de Mpro del SARS-CoV en palillos de color amarillo y del SARS-CoV-2 en palillos de color cyan; se muestra en rosado el inhibidor N3. Los residuos marcados con (*) corresponden al SARS-CoV-2.
Las poses determinadas para XNI, RTP y JT1 en RdRp del SARS-CoV-2 son altamente similares a aquellas observadas en los complejos XNI-HCV RdRp, JT1-HCV RdRp y RTP-HFMD RdRp. No obstante, se debe recordar que las enzimas RdRp del HCV y HFMD mantienen alrededor de un 46% de homología estructural con la polimerasa del SARS-CoV-2 y menos de 9% de homología de secuencia. Estos porcentajes de similitud se traducen en alteraciones en la configuración de los bolsillos, y consecuentemente en el modo de unión de los ligandos en la nsp12 con respecto a sus targets originales. Las Figuras 5A, 5B y 5C muestran la diferencia entre la posición original del ligando sometido a cribado y las posiciones adoptadas durante el acoplamiento al sitio de unión en RdRp.
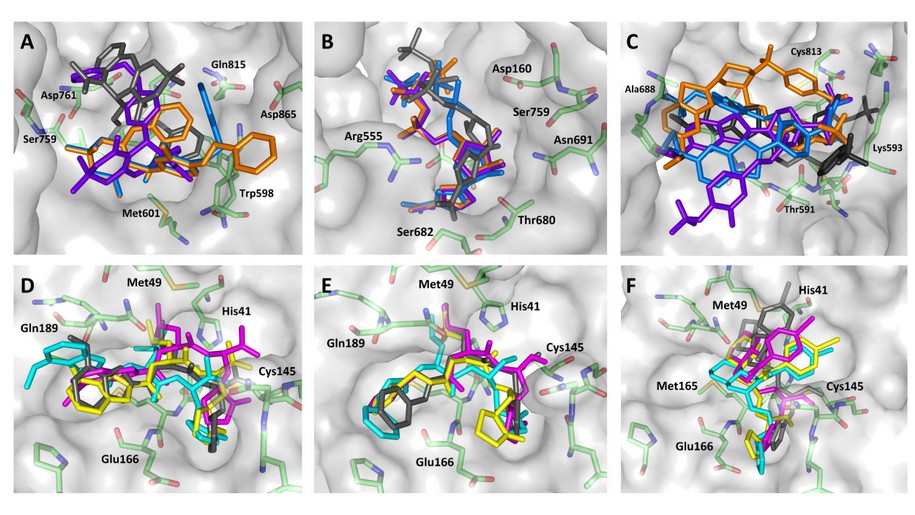
Figura 5. Posiciones adoptadas por los inhibidores de las enzimas del SARS-CoV-2 según el acoplamiento molecular. A) XNI-
RdRp. B) RTP-RdRp. C) JT1-RdRp. D) V3D-Mpro. E) EOF-Mpro. F) 3A7-Mpro. Se muestra en palillos de color negro la posición original del ligando y las mejores soluciones de cada resultado están representadas de diferentes colores. La proteína se muestra en superficie de color gris y los aminoácidos que interactúan directamente con los ligandos en el sitio de unión aparecen en palillos de color verde claro.
En cuanto al acoplamiento molecular para Mpro, se ensayaron 41 inhibidores en el sitio activo de la enzima. Los valores de afinidad de unión oscilaron entre -6 y -8 kcal/mol (Tabla Suplementaria 3). El mejor resultado se obtuvo con el ligando V3D, seguido por EOF y 3A7, con valores de afinidad de unión de -8, -7.9 y -7.8 kcal/mol, respectivamente, como se indica en la Tabla 2. Todos estos ligandos son inhibidores de la proteasa Mpro del SARS-CoV y sus estructuras químicas se muestran en la Figura 2. V3D es un inhibidor irreversible con un IC50 = 53 ± 1 µM. Por otra parte, EOF y 3A7 son inhibidores reversibles con valores de IC50 de 0.83 (EOF) y 63 µM (3A7). Como se mencionó anteriormente, las enzimas Mpro del SARS-CoV y SARS-CoV-2 son estructuralmente idénticas (SSE = 100%) y presentan una identidad de secuencia del 96.1%. En cuanto a sus sitios activos, el alineamiento de secuencias muestra la presencia de solo una sustitución (Arg188Ile) en Mpro del SARS-CoV, mientras que el resto de los residuos que contornean el sitio activo se mantienen conservados en ambas enzimas (Figura Suplementaria 2). La superposición de sitios activos de las enzimas Mpro del SARS-CoV y SARS-CoV-2 (Figura 4C) muestra que no solamente existe una alta homología de sus estructuras secundarias, sino que también se encuentran conservadas las conformaciones que adoptan los residuos que contornean su sitio de unión a ligandos de cada enzima. Esta elevada similitud explica por qué los inhibidores de Mpro del SARS-CoV son altamente afines a su enzima homóloga en SARS-CoV-2 en comparación a ligandos provenientes de otras proteasas, así como también explica la razón por la cual la pose adoptada por V3D, EOF y 3A7 en Mpro del SARS-CoV-2 es prácticamente igual a la pose que adoptan estos ligandos en las estructuras cristalinas de Mpro del SARS-CoV (Figura 5D a 5F).
Modo de unión de los complejos RdRp-RTP y Mpro-V3D
El análisis del modo de unión de RTP indica que este inhibidor es capaz de unirse a la RdRp del SARS-CoV-2 por medio de interacciones con 17 residuos de aminoácidos del sitio activo (Figura 6). Entre los más importantes se encuentran los residuos Arg624, Arg553 y Arg555, pues son capaces de generar múltiples interacciones de tipo puente salino y puente de hidrógeno con los átomos de oxígeno de la porción trifosfato de RTP. En el caso de Arg555, además del puente salino, la cadena lateral forma 3 puentes de hidrógeno con dos oxígenos de la porción trifosfato y uno del anillo de D-ribosa. Los residuos cargados Lys551 y Lys621 también generan puentes salinos con el trifosfato de RTP, mientras que los aminoácidos Asn691, Tyr680 y Tyr456 forman puentes de hidrógeno con la ribosa, el anillo de triazol y el grupo carboxamida, respectivamente. Finalmente, los residuos Thr556, Val557, Cys622, Asp623, Ser681, Ser682, Thr687, Ser759 y Asp760 asisten a la unión del inhibidor mediante enlaces débiles de Van der Waals.
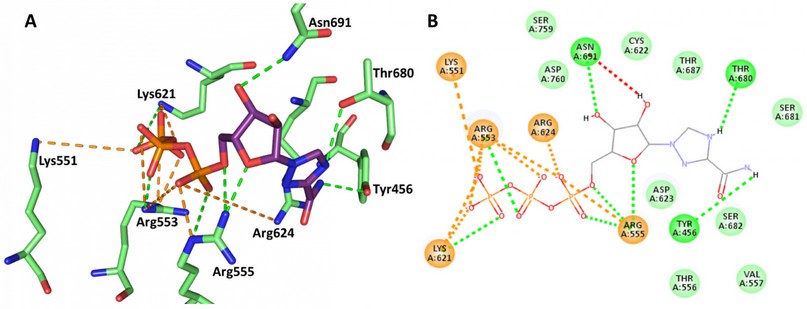
Figura 6. Interacciones entre la enzima RdRp (PDB ID: 7bv2) del SARS-CoV-2 y RTP en el sitio activo. A) Representación 3D. B) Representación 2D. Los puentes salinos y cargas atractivas se muestran en color naranja, los puentes de hidrógeno en color verde y las interacciones donante-donante desfavorables en color rojo.
El análisis del modo de unión de V3D en Mpro del SARS-CoV-2 muestra que el modo de unión del inhibidor es prácticamente igual a cuando se une a Mpro del SARS-CoV. El anillo de pirrol de la porción indol puede formar un enlace pi-alquil con Met165 y un puente de hidrógeno con Gln189 (Figura 7). Los grupos amino y carbonilo unidos al C-12 forman enlaces de hidrógeno con Gln189 y Glu166, respectivamente. Por su parte, el grupo carbonilo del C-34 forma un puente de hidrógeno con Gly143 mientras que otro puente de hidrógeno se forma entre el H del grupo amino en la porción de la oxopirrolidina y el residuo de Leu141. Adicionalmente, el oxígeno de la oxopirrolidina forma dos puentes de hidrógeno con la cisteína catalítica (Cys 145) y con Ser144. El segundo residuo catalítico, His41, es capaz de formar un enlace pi-catión con el grupo 3S-butil que se une al C-15 y un enlace Pi-alquil con el mismo grupo. Se observan además dos enlaces pi-alquil entre Met49 y 3S-butil. El grupo metoxi del anillo bencénico perteneciente al grupo forma dos enlaces pi-alquil con Leu167 y Pro168. También se observa numerosos enlaces carbono-hidrógeno entre V3D y los residuos de Thr25, Leu27, Tyr54, Phe140, Asn142, His163, His164, Asp187, Arg188, Thr190 y Gln192. Este modo de unión difiere significativamente del modo de unión de otros inhibidores de Mpro del SARS-CoV-2, como N3. La estructura cristalina del complejo N3-Mpro del SARS-CoV-2 muestra que el inhibidor se une al sitio activo con una conformación extendida. La cadena principal del inhibidor forma una hoja antiparalela con los residuos 164-168 de la cadena larga de la proteína, y con los residuos 189-191 del bucle que une a los dominios II y III. La Cys145 juega un rol fundamental, ya que es capaz de formar un enlace covalente con el ligando. Las cadenas laterales de Phe140, Asn142, Glu166, His163, His172 y las cadenas principales de Phe140 y Leu141, conforman el denominado subsitio S1, donde la His163 además crea un puente de hidrógeno con el anillo lactama de N3. En el subsitio hidrofílico S2 se incluyen las cadenas laterales de His41, Met49 y el residuo Leu27 en la región más profunda de la cavidad. Mientras que en el sitio P4, principalmente las cadenas laterales de Met165, Glu192 y Gln189 forman un pequeño bolsillo hidrofóbico. Finalmente, el sitio P5 es capaz de establecer interacciones de tipo Van der Waals con residuos como Pro168 y Thr19025, 47.
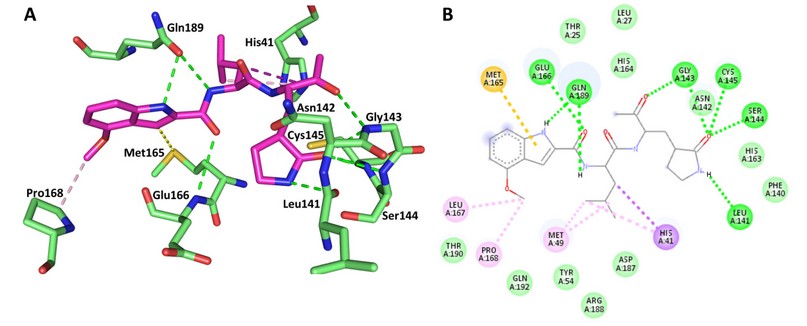
Figura 7. Interacciones entre la enzima Mpro (PDB ID: 7bqy) del SARS-CoV-2 y V3D en el sitio activo. A) Representación 3D. B) Representación 2D. Los enlaces de hidrógeno convencionales y enlaces carbono-hidrógeno se muestran en verde, el enlace Pi-alil en amarillo, los enlaces Pi-amida apilados y Pi-Alquil se muestran en rosado, y el enlace pi-catión en lila.
CONCLUSIONES
El análisis de homología realizado para enzimas RdRp y Mpro del virus causante del COVID-19 reveló que existen organismos virales que comparten casi el 50% de identidad estructural con la RdRp y hasta el 100% de identidad estructural con Mpro. El posterior alineamiento múltiple de secuencias permitió determinar que el sitio activo de la polimerasa del SARS-CoV-2 está altamente conservado entre organismos a pesar de la diferencia de tamaño entre enzimas. Por su parte, en Mpro también se determinó la conservación del sitio activo enzimático, aunque se han registrado unas pocas mutaciones puntuales. La alta conservación de sus sitios activos, que corresponden los principales bolsillos de unión de inhibidores, se debe al rol central que cumplen ambas enzimas en el ciclo de vida del virus. Por lo tanto, se pudo emplear el enfoque de redireccionamiento de fármacos que inicialmente tenían como blanco a las enzimas de organismos homólogos, para realizar un cribado virtual que permita identificar potenciales inhibidores contra el virus SARS-CoV-2.
Se llevaron a cabo ensayos de acoplamiento molecular en los que se pudo identificar a RTP y V3D como potenciales inhibidores de RdRp y Mpro, respectivamente. Ambas moléculas presentan valores de afinidad de unión inferiores a aquellas determinadas para RMP y N3, los cuales son moléculas con potente actividad in-vitro e in-vivo contra el SARS-CoV-2. Esto sugiere que RTP y V3D poseen un gran potencial de convertirse en opciones terapéuticas para combatir el SARS-CoV-2.
AGRADECIMIENTOS
El presente estudio se realizó gracias al auspicio de la Universidad Técnica de Ambato, a través de la Dirección de Investigación y Desarrollo, mediante el proyecto de investigación UTA-CONIN-2020-0295-R.
CONFLICTO DE INTERÉS
Los autores declaran no tener conflicto de interés.
REFERENCIAS
1. Pan American Health Organization/World Health Organization. Epidemiological Update: Coronavirus Disease, (2020, accessed 09/02/2021 2021).
2. World Health Organization. WHO Coronavirus (COVID-19) Dashboard. 2020.
3. Gorbalenya AE, Baker SC, Baric RS, et al. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. 2020; 5: 536.
4. Zhang L, Shen F-m, Chen F, et al. Origin and evolution of the 2019 novel coronavirus. 2020; 71: 882-883.
5. Helmy YA, Fawzy M, Elaswad A, et al. The COVID-19 pandemic: a comprehensive review of taxonomy, genetics, epidemiology, diagnosis, treatment, and control. 2020; 9: 1225.
6. Rangan R, Zheludev IN and Das RJB. RNA genome conservation and secondary structure in SARS-CoV-2 and SARS-related viruses. 2020.
7. Khailany RA, Safdar M and Ozaslan MJGr. Genomic characterization of a novel SARS-CoV-2. 2020; 19: 100682.
8. Gordon DE, Jang GM, Bouhaddou M, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. 2020; 583: 459-468.
9. Sanders JM, Monogue ML, Jodlowski TZ, et al. Pharmacologic treatments for coronavirus disease 2019 (COVID-19): a review. 2020; 323: 1824-1836.
10. Yin W, Mao C, Luan X, et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Sience 2020; 368: 1499-1504. DOI: 10.1126/science.abc1560.
11. Pastrian-Soto GJIjoo. Bases genéticas y moleculares del COVID-19 (SARS-CoV-2). Mecanismos de patogénesis y de respuesta inmune. 2020; 14: 331-337.
12. Joshi RS, Jagdale SS, Bansode SB, et al. Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. 2020: 1-16.
13. Báez-Santos YM, John SES and Mesecar AD. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. 2015; 115: 21-38.
14. Liu C, Zhou Q, Li Y, et al. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Publications, 2020.
15. Ju J, Li X, Kumar S, et al. Nucleotide analogues as inhibitors of SARS‐CoV polymerase. Pharmacology Research & Perspectives 2020; 8: e00674. DOI: 10.1002/prp2.674.
16. Shilatifard A. COVID-19: Rescue by transcriptional inhibition. American Association for the Advancement of Science, 2020.
17. Ullrich S and Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorganic & Medicinal Chemistry Letters 2020; 30. DOI: 10.1016/j.bmcl.2020.127377.
18. Hashemian SM, Farhadi T, Velayati AAJDd, development, et al. A review on remdesivir: a possible promising agent for the treatment of COVID-19. 2020; 14: 3215.
19. OMS. 10 Vaccines Granted Emergency Use Listing (EUL) by WHO. 2022. Organización Mundial de la Salud.
20. Knoll MD and Wonodi CJTL. Oxford–AstraZeneca COVID-19 vaccine efficacy. 2021; 397: 72-74.
21. Kim JH, Marks F and Clemens JDJNm. Looking beyond COVID-19 vaccine phase 3 trials. 2021: 1-7.
22. Gautret P, Lagier J-C, Parola P, et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. 2020; 56: 105949.
23. Dhama K, Sharun K, Tiwari R, et al. COVID-19, an emerging coronavirus infection: advances and prospects in designing and developing vaccines, immunotherapeutics, and therapeutics. 2020; 16: 1232-1238.
24. Lamb YN. Remdesivir: first approval. Springer 2020: 1-9. DOI: https://doi.org/10.6084/m9.figshare.12752432/.
25. Jin Z, Du X, Xu Y, et al. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. 2020; 582: 289-293.
26. Choudhary S, Malik YS and Tomar SJFii. Identification of SARS-CoV-2 cell entry inhibitors by drug repurposing using in silico structure-based virtual screening approach. 2020; 11: 1664.
27. Śledź P and Caflisch AJCoisb. Protein structure-based drug design: from docking to molecular dynamics. 2018; 48: 93-102.
28. Gupta SP. Chapter 3 - Inhibition of Viruses: Promising Targets and Their Importance. In: Gupta SP (ed) Studies on Hepatitis Viruses. Academic Press, 2018, pp.35-65.
29. Pagadala NS, Syed K and Tuszynski JJBr. Software for molecular docking: a review. 2017; 9: 91-102.
30. Krissinel E and Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallographica Section D: Biological Crystallography 2004; 60: 2256-2268.
31. Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic acids research 2000; 28: 235-242.
32. Schrödinger L. The PyMOL Molecular Graphics System. 2017.
33. Emsley P, Lohkamp B, Scott WG, et al. Features and development of Coot. Acta Crystallographica Section D 2010; 66: 486-501. DOI: doi:10.1107/S0907444910007493.
34. Di Tommaso P, Moretti S, Xenarios I, et al. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic acids research 2011; 39: W13-W17.
35. Artimo P, Jonnalagedda M, Arnold K, et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Research 2012; 40: W597-W603. DOI: 10.1093/nar/gks400
36. Forli S, Huey R, Pique ME, et al. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nature protocols 2016; 11: 905-919.
37. Trott O and Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry 2010; 31: 455-461.
38. Dallakyan S and Olson AJ. Small-molecule library screening by docking with PyRx. Chemical biology. Springer, 2015, pp.243-250.
39. Biovia DSJR. Materials Studio. 2017.
40. Venkataraman S, Prasad BV and Selvarajan R. RNA dependent RNA polymerases: insights from structure, function and evolution. Viruses 2018; 10: 76.
41. Chuanjun S, Xuan H, Ting H, et al. Potential inhibitors for targeting Mpro and Spike of SARS-CoV-2 based on sequence and structural pharmacology analysis. STEMedicine 2020; 1. DOI: 10.37175/stemedicine.v1i2.41.
42. Ugwu DI, Okoro UC and Ahmad H. New carboxamide derivatives bearing benzenesulphonamide as a selective COX-II inhibitor: design, synthesis and structure-activity relationship. PloS one 2017; 12: e0183807. DOI: 10.1371/journal.pone.0183807.
43. Matošević A and Bosak A. Carbamate group as structural motif in drugs: A review of carbamate derivatives used as therapeutic agents. Archives of Industrial Hygiene Toxicology 2020; 71: 285. DOI: 10.2478/aiht-2020-71-3466.
44. Ghosh AK and Brindisi M. Organic carbamates in drug design and medicinal chemistry. Journal of Medicinal Chemistry 2015; 58: 2895-2940. DOI: 10.1021/jm501371s.
45. Hang JQ, Yang Y, Harris SF, et al. Slow binding inhibition and mechanism of resistance of non-nucleoside polymerase inhibitors of hepatitis C virus. Journal of Biological Chemistry 2009; 284: 15517-15529. DOI: 10.1074/jbc.M808889200.
46. Hillen HS, Kokic G, Farnung L, et al. Structure of replicating SARS-CoV-2 polymerase. Nature 2020; 584: 154-156. DOI: 10.1038/s41586-020-2368-8.
47. Dai W, Zhang B, Jiang X-M, et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. 2020; 368: 1331-1335.
48. El‐Baba TJ, Lutomski CA, Kantsadi AL, et al. Allosteric Inhibition of the SARS‐CoV‐2 Main Protease: Insights from Mass Spectrometry Based Assays. Angewandte Chemie International Edition 2020; 59: 23544-23548. DOI: doi.org/10.1002/anie.202010316.
49. Gross AE and Bryson ML. Oral ribavirin for the treatment of noninfluenza respiratory viral infections: a systematic review. Annals of Pharmacotherapy 2015; 49: 1125-1135. DOI: https://doi.org/10.1177/1060028015597449.
Received: October 17, 2022 / Accepted: January 30, 2023 / Published:15 February 2023
Citation: Espín-Sánchez D, . Ramos-AristimbayM L, Sánchez-VacA S., Jaramillo-Guapisaca K, Vizueta-Rubio C, Chico-Terán F, Cerda-Mejía L, García M D. Identificación de inhibidores de las enzimas RdRp y Mpro del virus SARS-CoV-2 mediante homología estructural. Revis Bionatura 2023;8 (1)26. http://dx.doi.org/10.21931/RB/2023.08.01.26