2023.08.02.12
Files > Volume 8 > Vol 8 No 2 2023
Caracterización de protocolos de ensayos clínicos de productos farmacéuticos radicados para evaluación regulatoria en Colombia: consideraciones bioéticas y metodológicas
Characterization of clinical trial protocols of pharmaceutical products filed for regulatory evaluation in Colombia: bioethical and methodological considerations

Carlos J. Bello-Gándara1,3* , Erika Vergara-Cano2, David Osorio-Gallardo2, Juanita M. Recalde-Miranda3
, Erika Vergara-Cano2, David Osorio-Gallardo2, Juanita M. Recalde-Miranda3
1 Centro de Biociencias SURA, Medellín, Colombia
2 Programa de Química Farmacéutica, Universidad Icesi, Cali, Colombia.
3 Departamento de Ciencias Farmacéuticas, Universidad Icesi, Cali, Colombia.
* Correspondence: [email protected]
Available from: http://dx.doi.org/10.21931/RB/2023.08.02.12
RESUMEN
Los ensayos clínicos son esenciales para la evaluación de la seguridad y eficacia de productos farmacéuticos. Regiones emergentes como Latinoamérica han tenido un aumento en la ejecución de este tipo de estudios, tradicionalmente concentrados en países desarrollados. El propósito de este trabajo fue identificar las principales características con relevancia metodológica y bioética de los protocolos de ensayos clínicos de medicamentos presentados en Colombia ante la agencia regulatoria nacional. Se realizó un estudio observacional descriptivo, con base en el consolidado de los protocolos radicados al INVIMA y la información de EudraCT. Se incluyeron 597 protocolos, en los que se destaca el diseño aleatorizado (83,1%) doble ciego (66,7%), controlados por placebo (57,1%) y grupos paralelos (64,5%). Las enfermedades en investigación más frecuentes fueron neoplasias (22,8%). Se observó una baja representación de poblaciones vulnerables específicas como mujeres embarazadas (0,7%) o sujetos en situación de emergencia (2,4%) y escasas formulaciones especiales para población pediátrica (3,0%). La caracterización evidencia similitudes respecto al contexto regional y sugiere factores relevantes para evaluar y planificar protocolos en centros de investigación clínica.
Palabras clave: Características del estudio; Desarrollo de medicamentos; Investigación farmacéutica; Ensayos clínicos como tema; Protocolos de ensayos clínicos como tema.
ABSTRACT
Clinical trials are essential for the evaluation of safety and efficacy of pharmaceutical products. Emerging regions such as Latin America have had an increase in the execution of this type of study, traditionally concentrated in developed countries. The purpose of this work was to identify the main characteristics with methodological and bioethical relevance of drug clinical trial protocols submitted in Colombia to the national regulatory agency. A descriptive observational study was carried out, based on the consolidation of protocols filed with INVIMA and the information from EudraCT. 597 protocols were included, in which the randomized (83,1%), double-blind (66,7%), placebo-controlled (57,1%), and parallel group (64,5%) design stands out. The most frequent diseases under investigation were neoplasms (22,8%). A low representation of specific vulnerable populations was evidenced, such as pregnant women (0.7%) or subjects in an emergency situation (2.4%) and few special formulations for pediatric population (3.0%). The characterization shows similarities concerning the regional context and suggests relevant factors for evaluating and planning protocols in clinical research centers.
Keywords: Study characteristics; Drug development; Pharmaceutical research; Clinical trials as topic; Clinical trial protocols as topic.
INTRODUCCIÓN
Los ensayos clínicos constituyen el diseño metodológico fundamental para la evaluación de diversas intervenciones en salud en seres humanos, destinadas principalmente al diagnóstico, profilaxis y tratamiento de diversas patologías1. En el desarrollo de nuevos productos farmacéuticos, los ensayos clínicos son esenciales para generar la evidencia requerida acerca de su seguridad y eficacia para sustentar la autorización de comercialización por parte de agencias sanitarias, lo cual conlleva la implementación de protocolos de investigación con diferentes propósitos y atributos2.
De acuerdo con los lineamientos internacionales, como la guía para las Buenas Prácticas Clínicas de la Conferencia Internacional de Armonización (ICH, por sus siglas en inglés) y la pautas de la Declaración SPIRIT 2013, el protocolo es un elemento que tiene una función clave en la planificación y conducción de los ensayos, por lo que deben contar con unas características estándar que contribuyan a garantizar la sistematicidad en la ejecución de actividades, la integridad de los datos y la protección de los participantes en la investigación3,4.
Esta estricta regulación en investigación de medicamentos ha aportado a la calidad de los ensayos clínicos a nivel global, los cuales históricamente se han concentrado principalmente en regiones de altos ingresos como Norteamérica o Europa, dada su capacidad instalada y la ubicación de las principales empresas farmacéuticas5. Sin embargo, en las últimas décadas la investigación clínica se ha extendido a otras regiones emergentes como Asia, África y Latinoamérica, debido a condiciones más favorables para el reclutamiento de sujetos y la reducción de costos6. Específicamente en Latinoamérica han aumentado progresivamente el número de estudios en países como Chile, Argentina, Perú y Colombia7-10.
No obstante, debido a los distintos entornos regulatorios, es posible que los ensayos clínicos realizados fuera de los países de altos ingresos presenten diferencias en diseño y realización5. Adicionalmente, el acceso a la información sobre las características de los protocolos presenta limitaciones en ciertas regiones, ya que algunos países como Colombia no poseen registros primarios nacionales de ensayos clínicos con los lineamientos internacionales para tal fin, que faciliten la disponibilidad pública a información respecto a sus propiedades principales y conducción11. En este aspecto cabe resaltar el desarrollo de la Agencia Europea de Medicamentos (EMA, por sus siglas en inglés), que desde 2004 posee un registro detallado de ensayos clínicos que cumple los estándares estipulados por la Organización Mundial de la Salud (OMS) y se articula con la Plataforma de Registros Internacionales de Ensayos Clínicos12.
Teniendo en cuenta lo anterior, el presente estudio tuvo como objetivo identificar las principales características metodológicas e implicaciones bioéticas de los ensayos clínicos de medicamentos radicados en Colombia ante la agencia sanitaria nacional INVIMA (Instituto Nacional de Vigilancia de Medicamentos y Alimentos) para su evaluación regulatoria, a través de los datos disponibles por parte de la EMA de estos protocolos.
MATERIALES Y MÉTODOS
Se realizó un estudio observacional descriptivo, para lo cual se consultó el listado consolidado de los protocolos de investigación de medicamentos y productos biológicos radicados para evaluación de la autoridad regulatoria sanitaria INVIMA desde el año 2014 hasta enero 2022 (disponible en https://www.invima.gov.co/). La información detallada de las características específicas de los estudios clínicos fue buscada y extraída de la Base de Datos de Ensayos Clínicos de las Autoridades Reguladoras de Medicamentos de la Unión Europea (EudraCT) de la EMA, la cual registra todos los ensayos clínicos de intervención con medicamentos en la Unión Europea a partir del 1 de mayo de 2004 (disponible en https://eudract.ema.europa.eu).
Los ensayos clínicos elegibles para análisis debían contar con número de radicado en el consolidado colombiano y con número EudraCT asignado en el registro europeo. Se excluyeron estudios cuya codificación no permitiera su búsqueda efectiva en EudraCT para consultar su información correspondiente.
Se recolectaron datos agrupados en los siguientes dominios: identificación del estudio, estado regulatorio, características del medicamento en investigación, clasificación sistema órgano (SOC, por sus siglas en inglés) de la enfermedad según la terminología MedDRA de la ICH, características de la metodología experimental y características de la población elegible. Para el análisis descriptivo de las variables cuantitativas se calculó la mediana y rango intercuartílico (RIC) dada su distribución no normal, mientras que las variables cualitativas se abordaron a través de frecuencias absolutas y porcentaje. Los análisis estadísticos se efectuaron en el programa Stata 15 (StataCorp LLC; College Station, Texas, Estados Unidos).
Los datos empleados en este estudio se encuentran disponibles públicamente por parte de las agencias regulatorias. No se utilizaron datos de sujetos ni se identificaron individualmente los protocolos o patrocinadores. Por lo tanto, el estudio estuvo exento de revisión por comité de ética en investigación.
RESULTADOS
Se extrajeron 726 estudios clínicos a partir del consolidado de protocolos de investigación con medicamentos y productos biológicos radicados para evaluación regulatoria. Se excluyeron 129 estudios clínicos, de los cuales 121 no se tenían registro EudraCT y 8 presentaban un número de registro EudraCT no rastreable. En total, se incluyeron para análisis 597 ensayos clínicos conformados por 528 aprobados (88,4%), 15 requeridos (2,5%), 30 en desistimiento (5,0%), 7 en evaluación (1,2%), 11 negados (1,8%) y 6 en declaración de abandono (1,0%).
Respecto a la clasificación de las enfermedades en investigación, las más frecuentes fueron las neoplasias benignas, malignas y no especificadas (n=136, 22,8%) seguido de los trastornos musculoesqueléticos y del tejido conjuntivo (n=80, 13,4%) y las infecciones e infestaciones (n=77, 12,9%), Además, se observó que 14,9% (n=89) corresponden a enfermedades raras (Tabla 1).
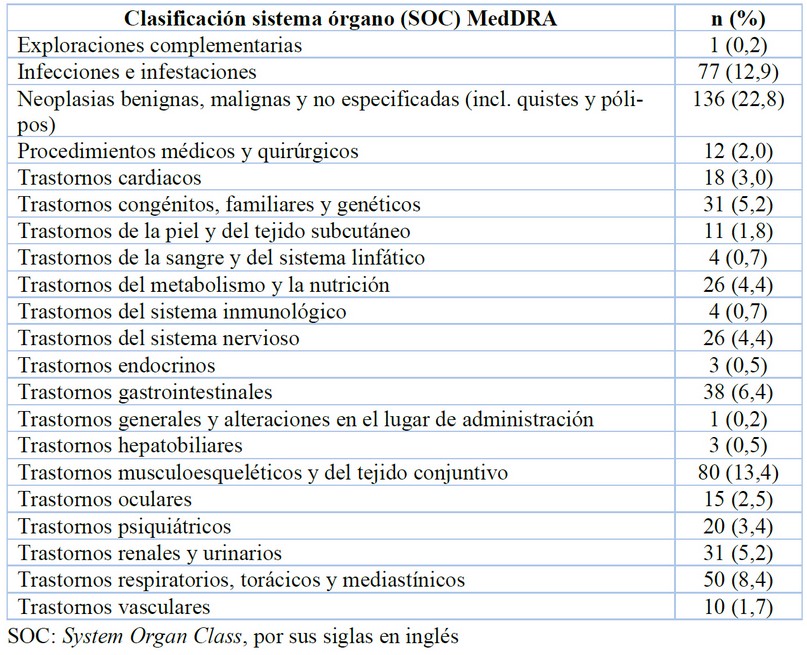
Tabla 1. Clasificación de las enfermedades en investigación
Así mismo, se encontró que entre las formas farmacéuticas de los medicamentos en investigación predominan las soluciones para inyección/infusión (n=210, 35,2%), seguido de las tabletas (n=181, 30,3%) y las cápsulas (n=58, 9,7%). Por otra parte, se observó una alta proporción de ingredientes farmacéuticos activos (IFA) de síntesis química (n= 326, 54,6%) seguido de IFA de origen biológico/biotecnológico (n= 269, 45,1%). Se resalta que 9,1% son catalogados como medicamentos huérfanos y 3,0% son formulaciones pediátricas (Tabla 2).

Tabla 2. Características de los medicamentos en investigación
Respecto al diseño de los protocolos de investigación (Tabla 3), la etapa de desarrollo clínico con mayor proporción es la fase III (79,6%). La mayoría de los estudios se caracterizan por ser aleatorizados (83,1%) doble ciego (66,7%), controlados por placebo (57,1%) y de grupos paralelos (64,5%). Un 67,0% incluye dentro de su alcance aspectos de farmacocinética y 25,3% contempla farmacogenética. Es importante enfatizar que 12,4% de los protocolos pertenecen a un plan de investigación pediátrico y 41% tiene resultados publicados en la base de datos. Adicionalmente, se estableció que la mediana del número de brazos de tratamiento en los estudios es 2 (RIC: 2-3) y tienen de 3 años (RIC: 2-5) de duración total, buscando reclutar una mediana de 580 sujetos (RIC: 252-1022) para la muestra total por protocolo, con variaciones de acuerdo con el grupo etario.

Tabla 3. Características del diseño de los protocolos de investigación
En las características de la población elegible, se evidenció que 84,8% de los protocolos incluyen participantes de edad adulta, mientras que 25,0% incluyen menores de 18 años. También se observa escasa presencia de las poblaciones vulnerables específicas que se evaluaron, entre las cuales la principal proporción se presenta en sujetos incapaces de dar su consentimiento personalmente (n= 103, 17,3%) (Tabla 4).
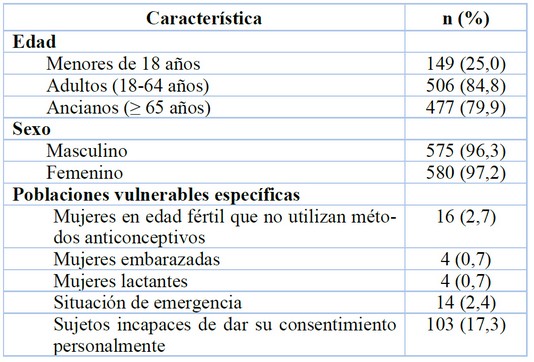
Tabla 4. Características de la población elegible en los ensayos clínicos
DISCUSIÓN
El presente estudio buscó identificar los principales atributos de los protocolos de ensayos clínicos de medicamentos que se radican ante la agencia sanitaria para su ejecución en Colombia. En primer lugar, se evidencia que las áreas de enfoque para desarrollo de los nuevos esfuerzos terapéuticos presentan estrecha similitud con las tendencias de investigación clínica a nivel regional. Aproximadamente, uno de cada cuatro estudios está relacionado con neoplasias y dicha categoría representa el área con mayor cantidad de estudios, lo cual ratifica el comportamiento reportado previamente para este grupo de patologías13.
Así mismo, se destacan también las investigaciones en infectología y trastornos musculoesqueléticos, estos últimos principalmente representados por condiciones reumatológicas. Por lo tanto, a nivel general los hallazgos están en línea con lo publicado previamente por otros autores en el contexto latinoamericano para países como Perú, Chile y Argentina7-9. Sin embargo, es notable la baja proporción de trastornos endocrinos, respiratorios y cardíacos observada en la muestra de protocolos, lo cual podría indicar una disminución de propensión hacia estas categorías frente a los resultados de Carreño y de Molina et al., en los cuales dichas áreas terapéuticas eran preponderantes en los ensayos clínicos en Colombia13,14.
Por otra parte, el estudio brinda un acercamiento a las características farmacéuticas de los productos involucrados en los ensayos clínicos. Se observa una amplia diversidad de formas farmacéuticas en las propuestas de investigación, con la principal proporción enfocada en soluciones parenterales y sólidos orales. En este ámbito, los productos parenterales ameritan especial atención por cuanto pueden implicar mayores requerimientos técnicos y logísticos por parte de los actores del proceso de investigación para garantizar el manejo integral del medicamento y la seguridad de los participantes15.
El estudio también denota el bajo porcentaje en formulaciones específicas para pacientes pediátricos. Esto constituye un indicio del limitado desarrollo de nuevas alternativas desde la fase de investigación para las necesidades especiales de los menores en aspectos como dosificación, administración y biodisponibilidad, implicando que en ciertos contextos de la práctica clínica se empleen formulaciones sin suficiente evidencia de uso en niños o se deba optar por preparaciones extemporáneas para pacientes individuales16,17. Además, la poca presencia de protocolos de terapias avanzadas señala otro campo de investigación con escasa incursión en el país con amplio potencial de crecimiento, lo cual incluye una variedad de abordajes novedosos de medicamentos que involucran terapia celular, terapia génica o ingeniería de tejidos.
Frente al origen del principio activo, se observa una paridad entre estudios con compuestos de síntesis química y aquellos con moléculas complejas de origen biológico o biotecnológico. Lo anterior puede explicarse por el fuerte enfoque global de la industria farmacéutica en éstos últimos, ya que representan un importante enfoque de investigación y desarrollo por sus diversas ventajas y aplicaciones terapéuticas en diferentes patologías18.
Al indagar las características del diseño metodológico de los ensayos clínicos, es posible evidenciar que en Colombia predominan ampliamente los protocolos de fase III aleatorizados, de dos grupos paralelos, doble ciego y controlados con placebo, lo cual es propio de los estudios pivotales que sustentan las solicitudes de autorización de nuevos medicamentos ante agencias regulatorias. Esto coincide con los hallazgos previos en Colombia y otros países latinoamericanos, donde dicho perfil era el más común para los estudios7-10. Así mismo, el número global de sujetos a enrolar en dichos protocolos es alto, lo que da cuenta que principalmente se radican estudios de confirmación terapéutica por encima de aquellos de fases exploratorias o de prueba de concepto que implican un menor tamaño de muestra.
Adicionalmente, en línea con las tendencias farmacotécnicas mencionadas, son escasos los protocolos que pertenecen formalmente a un plan de investigación pediátrico y se observa que solo un cuarto del total de protocolos contempla la inclusión de menores de 18 años. En dicho grupo etario, la mediana de la meta de enrolamiento fue sustancialmente menor que para otras edades, aspecto ligado a las consideraciones éticas especiales que influyen en el reclutamiento y retención de estos participantes.
Respecto a la población elegible, es destacable la reducida cantidad de estudios que incluyen alguna de poblaciones vulnerables que fueron evaluadas, especialmente aquellos relacionados con mujeres en estado de embarazo y lactantes. Esto refuerza la reflexión planteada por el Foro Global de Bioética en Investigación respecto a las limitaciones para incluir este grupo específico, lo que genera como consecuencia la dificultad que afrontan los profesionales de la salud para encontrar evidencia sobre la eficacia y seguridad de los medicamentos en dicha población19. Por lo tanto, resulta necesario promover la investigación farmacéutica que incluya mujeres embarazadas y otras poblaciones sistemáticamente excluidas de los estudios, garantizando las medidas especiales de protección y minimización de riesgos, además de actualizar de manera oportuna los paradigmas éticos y la normatividad asociada20.
CONCLUSIONES
La perspectiva obtenida a través del registro EudraCT acerca de las características metodológicas de los protocolos de ensayos clínicos de medicamentos en Colombia da cuenta de similitudes respecto a las tendencias del contexto regional latinoamericano, permitiendo identificar que predomina la investigación en neoplasias y estudios de confirmación terapéutica en fase 3 doble ciego controlados con placebo, presentando una gran diversidad de formas farmacéuticas y tipos de IFA respecto a los productos en investigación. Así mismo, desde el ámbito bioético es pertinente resaltar la escasa inclusión de poblaciones vulnerables específicas en los protocolos junto a la poca investigación de formulaciones pediátricas y medicamentos huérfanos como factores sustanciales que deben optimizarse para mejorar la generación de evidencia clínica de productos farmacéuticos.
Financiamiento: El estudio fue autofinanciado.
Conflicto de interés: Los autores declaran no tener conflictos de interés.
Contribuciones de los autores: CJBG y JMRM realizaron la conceptualización, diseño del estudio y gestión de la investigación. CJBG, DOG, EVC fueron responsables de la recolección de datos. CJBG, DOG, EVC y JMRM desarrollaron el análisis e interpretación de datos, así como en la redacción, revisión y aprobación de la versión final del artículo. Todos los autores asumen la responsabilidad por el artículo.
REFERENCIAS
1. Umscheid CA, Margolis DJ, Grossman CE. Key concepts of clinical trials: A narrative review. Postgrad Med. 2011;123(5):194–204. doi: 10.3810/pgm.2011.09.2475.
2. Fruhner K, Hartmann G, Sudhop T. Analysis of integrated clinical trial protocols in early phases of medicinal product development. Eur J Clin Pharmacol. 2017;73(12):1565–77. doi: 10.1007/s00228-017-2335-y.
3. Chan AW, Tetzlaff JM, Altman DG, Laupacis A, Gøtzsche PC, Krleža-Jeric K, et al. Declaración SPIRIT 2013: definición de los elementos estándares del protocolo de un ensayo clínico. Revista Panamericana de Salud Pública. 2015;38(6):506–14.
4. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). ICH Harmonised Guideline Integrated Addendum to ICH E6(R1): Guideline for Good Clinical Practice E6(R2) [Internet]. 2016 [citado el 26 de mayo de 2022]. Disponible en: https://database.ich.org/sites/default/files/E6_R2_Addendum.pdf
5. Murthy S, Mandl KD, Bourgeois FT. Industry-sponsored clinical research outside high-income countries: An empirical analysis of registered clinical trials from 2006 to 2013. Health Res Policy Syst. 2015;13:28. doi: 10.1186/s12961-015-0019-6.
6. Jeong S, Sohn M, Kim JH, Ko M, Seo H won, Song YK, et al. Current globalization of drug interventional clinical trials: Characteristics and associated factors, 2011-2013. Trials. 2017;18:288. doi: 10.1186/s13063-017-2025-1.
7. Traversi L, Bolaños R. Analysis of clinical drug trials in children compared to clinical drug trials in adults, in Argentina. Arch Argent Pediatr. 2019;117(1):34–40. doi: 0.5546/aap.2019.eng.34.
8. Aguilera B. Trends in clinical trials performed in Chile. Rev Med Chil. 2021;149:110–8. doi: 10.4067/S0034-98872021000100110
9. Minaya G, Fuentes D, Obregón C, Ayala-Quintanilla B, Yagui M. Características de los ensayos clínicos autorizados en el Perú, 1995-2012. Rev Peru Med Exp Salud Publica. 2012;29(4):431–6. doi: 10.17843/rpmesp.2012.294.385.
10. Molina de Salazar DI, Álvarez-Mejía M. Estado de la investigación clínica en Colombia. Acta Médica Colombiana. 2018;43(4):179–82. doi: 10.36104/amc.2018.1374.
11. Lemmens T, Herrera Vacaflor C. Transparencia sobre los ensayos clínicos en la Región de las Américas: necesidad de coordinar las esferas regulatorias. Revista Panamericana de Salud Pública. 2019;43:1–7. doi: 10.26633/rpsp.2019.303.
12. Venugopal N, Saberwal G. A comparative analysis of important public clinical trial registries, and a proposal for an interim ideal one. PLoS One. 2021;16(5):e0251191 doi: 10.1371/journal.pone.0251191.
13. Carreño Dueñas A. Situación de los ensayos clínicos en Colombia. Medicina. 2013;35(2):123–9.
14. Molina de Salazar DI, Botero SM, Giraldo GC. Investigación clínica y ensayos clínicos ¿En qué vamos? Acta Médica Colombiana. 2016;41(3S):43–50.
15. de Camargo Silva AEB, Moreira Reis AM, Inocenti Miasso A, Oliveira Santos J, de Bortoli Cassiani SH. Eventos adversos causados por medicamentos en un hospital centinela del Estado de Goiás, Brasil. Rev Lat Am Enfermagem. 2011;19(2):1–9. doi: 10.1590/S0104-11692011000200021.
16. Sánchez-González E. ¿Qué sabe ud. acerca de formulaciones pediátricas? Revista Mexicana de Ciencias Farmacéuticas. 2015;46(2):68–70.
17. González C. Farmacología del paciente pediátrico. Revista Médica Clínica Las Condes. 2016;27(5):652–9. doi: 10.1016/j.rmclc.2016.09.010.
18. Barrera LA. Desarrollo de medicamentos biotecnológicos. Del laboratorio al paciente. Medicina. 2018;40(1):44–55.
19. Saenz C, Alger J, Beca JP, Belizán JM, Cafferata ML, Arturo J, et al. Un llamado ético a la inclusión de mujeres embarazadas en investigación: Reflexiones del Foro Global de Bioética en Investigación. Revista Panamericana de Salud Pública. 2017;41:e13. doi: 10.26633/RPSP.2017.13.
20. Carracedo S, Palmero A, Neil M, Hasan-Granier A, Saenz C, Reveiz L. El panorama de los ensayos clínicos sobre COVID-19 en América Latina y el Caribe: evaluación y desafíos. Revista Panamericana de Salud Pública. 2021;45:e33. doi: 10.26633/RPSP.2021.33.
Received: 2 January 2023/ Accepted: 19 April 2023 / Published:15 June 2023
Citation: Bello-Gándara C J, Vergara-Cano E, Osorio-Gallardo D, Recalde-Miranda J M. Caracterización de protocolos de ensayos clínicos de productos farmacéuticos radicados para evaluación regulatoria en Colombia: consideraciones bioéticas y metodológicas. Revis Bionatura 2023;8 (2) 12. http://dx.doi.org/10.21931/RB/2023.08.02.12