Enfermedad Poliquistica
Files > Volume 1 > Vol 1 No 1 2016 > Revisiones

Enfermedad Renal Poliquística
Polycystic Kidney Disease
Available from: http://dx.doi.org/10.21931/RB/2016.01.01.7
Pedro Cena Rivero1, Rocío Elizabeth Castillo Andrade2, Silvia Marcela Baquero3, Viviana Margarita Espirel 4
RESUMEN
La Enfermedad Renal Poliquística es una enfermedad genética conocida por sus siglas en Inglés PKD (Polycystic Kidney Disease) y se caracteriza por la aparición progresiva de lesiones quísticas en los riñones que sustituyen el parénquima renal, causando el deterioro de su función hasta el estadio 5. La PKD constituye una de las causas de Enfermedad Renal Crónica bajo tratamiento sustitutivo renal (TSR). Tiene dos patrones de herencia: el patrón autosómico dominante y el patrón autosómico recesivo. La forma dominante es más común pero menos grave que la forma recesiva. Se conoce que es causada por la mutación en varios loci del genoma humano. La forma autosómica dominante puede ser causada por mutaciones en dos genes diferentes (PKD1 y PKD2), a diferencia de la forma autosómica recesiva que sólo tiene un gen causal (PKHD1). En la actualidad la comunidad científica dirige esfuerzos hacia el conocimiento más profundo de la fisiopatología de esta entidad que permita desarrollar alternativas terapéuticas encaminadas a retrasar la aparición de los quistes o la progresión de los que ya están instaurados. El objetivo es sistematizar el conocimiento científico disponible acerca de la Enfermedad Renal Poliquística, y brindar un medio de consulta actualizada sobre las características clínicas y terapéuticas de la misma.
Palabras clave: Enfermedad renal poliquística; enfermedad renal crónica; patrón autosómico dominante; patrón autosómico recesivo.
ABSTRACT
The Polycystic Kidney Disease (PKD) is a genetic disease which is characterized by the gradual emergence of cystic lesions in the kidneys, which replace the renal parenchyma causing deterioration of its function to stage 5. The PKD is one of the causes of Chronic Kidney Disease on renal replacement therapy (RRT). The Polycystic Kidney Disease has two patterns of inheritance: autosomal dominant pattern and the autosomal recessive pattern. The dominant form is more common but less severe than autosomal recessive form. PKD is known that is caused by mutations in several loci of the human genome. The autosomal dominant form can be caused by mutations in two different genes (PKD1 and PKD2), unlike the autosomal recessive form only has a causal gene (PKHD1). At present the international scientific community efforts toward deeper understanding of the pathophysiology of this entity for the purpose of developing therapeutic alternatives that avoid the appearance of cysts or progression of those already in place. The aim is to systematize the available scientific knowledge about Polycystic Kidney Disease and provide a source of consultation update on clinical characteristics and therapeutic options for patients with PKD.
Keywords: Polycystic kidney disease, chronic kidney disease, autosomal dominant, autosomal recessive pattern.
Introducción
La enfermedad renal poliquística es conocida por sus siglas en inglés como PKD (Policistyc Kidney Disease) y se caracteriza por el desarrollo de quistes epiteliales en ambos riñones, los que van aumentando progresivamente en tamaño y en número, hasta desplazar el parénquima, con la consecuente aparición de la falla funcional. Es una enfermedad de origen genético, multisistémica, de inicio tardío en la forma autosómica dominante y de inicio temprano si es recesiva.
La PKD se produce por mutaciones en varios loci humanos. La forma autosómica dominante la causan mutaciones en dos genes diferentes: PKD1 y PKD2. El 85 % de los individuos con enfermedad renal poliquística autosómica dominante (ADPKD) tiene mutaciones en el gen PKD1, mientras que el 15 % restante posee mutaciones en el gen PKD2.
Las principales características patogénicas son el aumento de la proliferación celular tubular y la formación de los quistes a lo largo de la nefrona.1 Estos pacientes suelen presentar además, quistes en otros órganos como el hígado, cerebro y páncreas. 2
La PKD muestra una considerable prevalencia a nivel mundial 3 y es la causa del 10 % de los pacientes que padecen enfermedad renal crónica en estadio 5, condición ésta que requiere de terapia sustitutiva renal, con los elevados costos que implica, por lo cual resulta de gran interés para la comunidad científica, la profilaxis de la PKD, así como hallar alternativas de tratamiento para los afectados, especialmente terapias que retrasen la aparición y el desarrollo de los quistes renales, como un modo de proteger el parénquima, mejorar la calidad de vida de los enfermos, y disminuir el impacto económico y social de la terapia sustitutiva renal, que en el Ecuador alcanzó en el año 2013, un total de 8 253 pacientes con este tratamiento.4 Si calculamos que aproximadamente el 10 % los pacientes con Enfermedad Renal Crónica (ERC) ha llegado la Terapia Sustitutiva Renal (TSR) como consecuencia de la PKD, tendríamos que su número asciende aproximadamente a 825 individuos. Tan sólo en la ciudad de Ibarra, 252 pacientes recibían TRS en el año 2013.4
Enfermedad Renal Poliquística Autosómica
Dominante
Prevalencia. La Enfermedad Renal Poliquística autosómica dominante, en inglés Autosomal Dominant-PKD (ADPKD) es la enfermedad renal hereditaria más frecuente. Se ha descrito que su prevalencia, alcanza 1/800 en todos los grupos étnicos y afecta 1/500-1/1000 recién nacidos vivos.6 Aproximadamente 2700 personas con ADPKD iniciaron terapia de reemplazo renal en el 2008 en los Estados Unidos de América, las que se añadieron a las 26.000 personas que ya recibían dicha terapia por causa de esta enfermedad.7
Si bien la ADPKD es una importante causa de enfermedad renal crónica en el adulto, la misma ha ganado importancia en Pediatría desde que se describieron los primeros casos en niños. Su diagnóstico prenatal en determinados casos, ya es posible desde hace más de 20 años. El reporte más temprano es a las 16 semanas de gestación. Se estima que aproximadamente el 2 % de los niños afectados se presentan con manifestaciones graves.
La inesperada aparición infantil de la enfermedad en una familia con antecedentes de inicio clásico en adultos, no tiene una explicación satisfactoria a pesar de que se reporta cierta variabilidad en su expresión familiar.8 Zerres y otros, estudiaron 64 familias con 79 niños afectados y diagnosticaron cinco casos prenatalmente en 17 familias con más de un hijo con la enfermedad.11 Se ha señalado que aproximadamente en el 30 % de las personas en riesgo, el diagnóstico puede establecerse por ultrasonografía antes de los 10 años de edad,18 aunque en el 40 % de los afectados no ha sido posible detectar quistes antes de los 30 años de edad.9
Fisiopatología
La ADPKD se caracteriza por el desarrollo de lesiones quísticas renales que afectan el funcionamiento de la nefrona, cuya primera manifestación clínica es la hipostenuria (pérdida de la capacidad de concentrar orina); posteriormente se reduce el flujo sanguíneo renal que desencadena en una pérdida progresiva del parénquima, que va siendo remplazado progresivamente por lesiones quísticas. Esto lleva a la aparición de complicaciones propias del daño renal como son la hipertensión arterial y la anemia. Asimismo, aumenta el riesgo de infecciones urinarias a repetición y la aparición de dolor en el dorso y los flancos como manifestación del aumento de tamaño quístico, su infección o sangrado interno.
La evolución progresiva de las lesiones renales conduce a la ERC grado 5, que a los 70 años de edad ya padece el 70 % de los afectados por ADPKD. En estos pacientes se observa además, la aparición de quistes en otros órganos, como el hígado, las vesículas seminales, el páncreas y la membrana aracnoides, así como alteraciones vasculares, dígase aneurismas intracraneales, dilatación del arco aórtico, disección de la aorta torácica, prolapso de la válvula mitral y hernias en la pared abdominal.10
Las manifestaciones clínicas están directamente relacionadas con el tamaño de los quistes y con el grado de afectación del parénquima. El volumen de los quistes aumenta de manera exponencial. Según estudios realizados, se conoce que el incremento de volumen en estos quistes es alrededor del 5,3 % al año.11 Como antes se expuso, la ADPKD puede ser causada por mutaciones en dos genes diferentes; la más frecuente es la ADPKD1 (85 % de los casos) que es causada por mutaciones en el gen PKD1 o también llamado policistina1. Este gen codifica la síntesis de una proteína del mismo nombre; la proteína policistina 1 que es una glicoproteína transmembrana, reguladora de canales calcio y de la homeostasis del calcio intracelular. También cumple un papel muy importante en la interacción célula-célula y célula-matriz.12 El 15 % de los pacientes restantes se deben a mutaciones en el gen PKD2, que codifica la proteína policistina 2, una proteína transmembrana que actúa como canal de calcio voltaje dependiente y regula el calcio intracelular, especialmente en el retículo endoplásmico. Esta proteína está implicada en el movimiento ciliar de las células epiteliales donde se expresa fenotípicamente.
Evolución de la enfermedad
Un estudio realizado por el Consorcio de Estudios Imagenológicos para valorar la progresión en la PKD (CRISP) realizó un seguimiento prospectivo a 214 individuos afectados, con imágenes anuales, y mostró que el volumen total renal y el volumen de los quistes aumentan de forma exponencial. El promedio del volumen renal total fue de 1060 ± 642 ml de línea de base, y se encontró que el incremento anuales de 204 ± 246 ml por año (5,27 ± 3,92% alaño), durante un periodo de seguimiento de 3 años. 13,3
La función renal de los pacientes con ADPKD se mantiene conservada hasta aproximadamente los 40 años de edad, a expensas de una hipertrofia compensatoria en las nefronas funcionantes.14 Pero la enfermedad continúa su progresión y alrededor de los 50 años ya se encuentra algún grado alteración en la tasa de filtración glomerular (TFG), la cual va disminuyendo con el transcurso del tiempo hasta llegar a un valor por debajo de 15 ml/min (ERC estadio 5).15 Los riñones pueden alcanzar un gran tamaño, aproximadamente 25 libras en una mujer de 1,55 m. Se considera que el 50 % de los individuos con ADPKD desarrollará ERC estadio 5 para a los 60 años. La edad de presentación de la falla renal es más temprana en PKD1 que en PKD2 (54 vs.74 años),3 sin embargo, la edad de inicio de la falla renal es variable, incluso dentro de la misma familia. Se ha determinado, que la ADPKD1 es más severa que la ADPKD2, ya que presenta un mayor aumento del tamaño renal, así como un mayor número de quistes. Se ha demostrado que la tasa de crecimiento de los quistes es similar para los dos tipos.16
El fenómeno de expresividad variable se pone de manifiesto con la aparición, en una misma familia, de casos graves y casos leves. La variabilidad fenotípica involucra diferencias en la tasa de filtración glomerular, la edad a la que se alcanzó la falla renal terminal, la ocurrencia de hipertensión arterial, la presencia de quistes extrarrenales sintomáticos y la hemorragia subaracnoidea por aneurismas intracraneales en frambuesa.17 Aunque esta es una enfermedad autosómica dominante, entre el 10 y el 20 % de los pacientes no tiene historia familiar previa, se trata de mutaciones de novo o posiblemente un infra diagnóstico de los familiares afectados. 18
Diagnóstico
El diagnóstico se establece inicialmente mediante estudios imagenológicos como ecografía renal o tomografía axial computarizada (TAC). Para el diagnóstico de la enfermedad poliquística se necesita la presencia de 3 a 5 quistes en cada riñón (figura 1).19 La sospecha clínica se basa en los hallazgos específicos en función de la edad. La ecografía renal es el método más usado por su bajo costo y fácil disponibilidad, el cual reporta riñones aumentados de tamaño y de contornos irregulares con múltiples quistes, aunque sólo detectan quistes mayores a 8 mm y se debe tener en cuenta que en individuos jóvenes o con PKD2, es posible que el informe ecográfico no los reporte y pasen inadvertidos.20,21
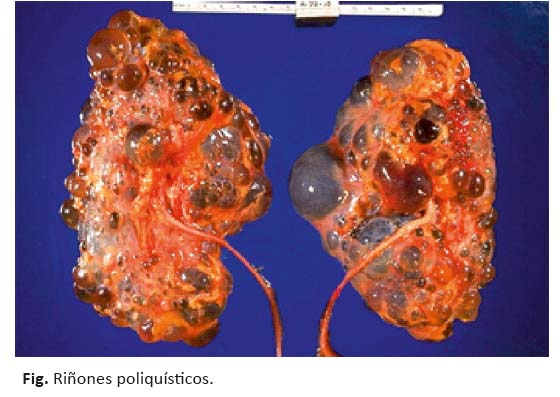
Se han realizado estudios para determinar la sensibilidad y especificidad de la ultrasonografía en el diagnóstico de la ADPKD tipos 1 y 2, comparado con el análisis de ligamiento. La sensibilidad de la ecografía renal para el diagnóstico fue comparada con los resultados de los genotipos inferidos por los estudios de ligamiento. La sensibilidad de la ecografía renal para diagnosticar la ADPKD, en menores a 30 años, fue del 95 % para ADPKD tipo1 pero sólo del 67 % para ADPKD tipo 2. Sin embargo, la sensibilidad de la ecografía, tanto para ADPKD tipo1 como tipo 2, es del 100 %.22,23
Tratamiento
El tratamiento se encamina a controlar las manifestaciones secundarias y a evitar el deterioro de la función renal por las comorbilidades asociadas, en particular el control de la Hipertensión Arterial, para lo cual se usan inhibidores de la enzima convertidora de angiotensina (IECA) o antagonistas de los receptores de la angiotensina II (ARA II), ya que han mostrado reducción en la microalbuminuria en individuos con PKD, efecto que no se ha encontrado con los bloqueadores de canales de Calcio. Se ha demostrado que la tensión arterial media debe ser menor a 92 mm Hg, ya que una mayor tensión arterial está relacionada con mayor mortalidad.24
Para el manejo del dolor, en primer lugar, se debe descartar infección, la presencia de litiasis o de tumor. Una vez descartadas se pondrá en práctica el tratamiento escalonado para el dolor, con la precaución de evitar o reducir a dosis mínimas, la medicación nefrotóxica. En determinados casos de difícil alivio se ha recomendado el uso de antidepresivos tricíclicos, los que han mostrado buenos resultados.3 Los agentes narcóticos se reservan para las crisis agudas, ya que su uso crónico produce dependencia física y psicológica. Cuando las medidas anteriores para el manejo del dolor crónico no han tenido éxito, se puede realizar bloqueo neural, aspiración del quiste o terapia esclerosante.9,25,29 Existen otras terapias quirúrgicas para el manejo del dolor, que van desde descompresión quirúrgica, penetración laparoscópica, denervación renal, hasta la nefrectomía.
Dentro de las medidas específicas para la nefroprotección, es importante recalcar las recomendaciones propias del paciente con Enfermedad Renal Crónica en sus diferentes estadios, como la indicación de una dieta con restricción de proteínas (a 0,6 g proteína/kg peso corporal/día), así como promover la ingesta hídrica mínima de 2000 ml/día.26
Asesoría genética
La mayoría de los individuos afectados con PKD autosómica dominante tiene, por lo menos, un familiar afectado. La incidencia de mutaciones de novo es significativa y ocurre en cerca del 10 % de las familias afectadas. Las recomendaciones para la evaluación de los familiares de un probando afectado, con una aparente mutación de novo, incluye un tamizaje adecuado por métodos imagenológicos, especialmente en PKD2. 28 Se debe señalar que la historia familiar puede parecer negativa, por falla en el diagnóstico, muerte temprana del individuo afectado antes del inicio de los síntomas, o inicio tardío de la enfermedad en el familiar afectado. En el caso del hermano de un afectado, el riesgo varía así:
• Si alguno de los padres está afectado, el riesgo en cada uno de sus hijos es del 50 %.
• Si ninguno de los padres tiene alteraciones en las imágenes renales, ni tampoco en el examen molecular, lo más probable es que se deba a una mutación de novo; por lo tanto, el riesgo que la enfermedad se repita en otros hijos es similar al de la población general.28
Consideraciones en la asesoría del trasplante renal
Es importante realizar una valoración muy estricta a los familiares de un paciente con ADPKD, ya que estos pueden también tener la enfermedad y aún no manifestarla. En este caso, se contraindica la donación del riñón.3 La evaluación para descartar la enfermedad tiene que ser molecular e imagenológica. Usualmente los pacientes con PKD ingresan a lista para donantes cadavéricos.30
Terapias bajo investigación
Los estudios realizados tanto en el modelo animal como en humanos con ADPKD indican que los quistes son responsables del declive en la tasa de filtración glomerular, que ocurre de manera tardía en el curso de la enfermedad.31 Esto se debe a la disrupción anatómica en la filtración glomerular y los mecanismos de concentración urinaria a escala masiva, asociados a la compresión y obstrucción de las nefronas adyacentes, por los quistes en la corteza, médula y papila. Los quistes dificultan el drenaje de orina, lo cual conduce a la atrofia tubular y pérdida de la función del parénquima renal, por mecanismos similares a la obstrucción ureteral. Estos producen citosinas y factores de crecimiento que resultan en fibrosis y falla renal.
Por lo tanto, es una estrategia razonable el prevenir o disminuir la progresión de formación de quistes, para evitar los cambios secundarios y así prolongar la función renal en estos pacientes.31 Se ha comprobado que la proteína mammalian target of rapamycin (mTOR) está activada de manera aberrante en el epitelio quístico de los pacientes con ADPKD.7 Por tal motivo, se han realizado estudios clínicos en humanos, para comprobar si medicamentos inhibidores de la mTOR, como el Sirolimus, presentan eficacia en detener la progresión de la enfermedad.18,31
El Sirolimus es un macrólido, que actualmente se usa como inmunosupresor en la prevención del rechazo de trasplante renal. Este medicamento fue descubierto inicialmente como un producto de la bacteria Streptomyces hygroscopicus. Su efecto inmunosupresor se basa en que inhibe la respuesta a la interleucina 2 (IL-2), bloqueando la activación de los linfocitos B y T. El Sirolimus se une a una proteína citoplasmática llamada FKBP12 y forma el complejo Sirolimus/FKBP12, el cual inhibe la vía del mTOR. Este complejo mTOR también es llamado FRAPo RAFT y juega un importante papel en el crecimiento y proliferación celular. 11,33
El Sirolimus ha mostrado beneficios significativos en el modelo murino para PKD y ha reducido la progresión de la enfermedad.19 Un estudio aleatorizado, controlado, con 18 meses de ingesta de Sirolimus en adultos con ADPKD y enfermedad renal crónica temprana no encontró evidencia de que el Sirolimus a dosis de 2 mg/día, enlenteciera el crecimiento renal poliquístico, comparado con el grupo control.17
En cuanto a otros inhibidores de la vía mTOR, tenemos al Everolimus, con el cual se realizado un ensayo aleatorizado, doble ciego, con 433 pacientes con ADPKD, que recibieron durante 2 años Everolimus a dosis de 2,5 mg dos veces al día vs. placebo en el grupo control. Los resultados de este estudio muestran que el Everolimus disminuyó el incremento en el volumen renal total de los pacientes con ADPKD, durante el primer año; pero no disminuyó la progresión del daño renal. Sin embargo el uso de Everolimus estuvo asociado a una elevada tasa de efectos adversos, similar a las encontradas en trasplante renal.34
Existen reportes de familias con ADPKD, que no presentan mutaciones en PKD1 ni en PKD2, por lo que se presume la existencia de otro gen relacionado (PKD3), aunque ello todavía no ha sido demostrado.2
Un mejor conocimiento de la fisiopatología y la disponibilidad de modelos animales han permitido el desarrollo de otros fármacos prometedores para ensayos clínicos.
Antagonistas de vasopresina
Estudios recientes en modelos animales han demostrado que la regulación de los niveles de AMPc por vía de los receptores V2 puede inhibir drásticamente el desarrollo de quistes renales.45,46
El consumo de una cantidad elevada de agua también ejerce un efecto protector por sí solo, en el desarrollo de poliquistosis renal en ratas; probablemente esto se debe a la inhibición de la secreción de vasopresina. Recientemente, se han completado estudios clínicos en fase II con tolvaptan (antagonista con alta potencia y selectividad para el receptor de vasopresina V2 humano) y un estudio en fase III se encuentra en marcha.47
Análogos de la somatostatina
La somatostatina actúa sobre los receptores SST2 e inhibe la acumulación de AMPc no sólo en el riñón sino también en el hígado. La octreotida, un análogo sintético de la somatostatina, ha demostrado causar un enlentecimiento en el aumento de tamaño de quistes renales y hepáticos en modelos animales de PQR y en riñones poliquísticos. (p) Tres recientes estudios prospectivos aleatorizados con control y octreotida o lanreotida durante 6 a 12 meses mostraron una reducción en el volumen hepático en pacientes con PQH y buena tolerancia. 47,48
Otras estrategias dirigidas contra los mecanismos moleculares que se encuentran alterados en la PKD han demostrado tener resultados prometedores en modelos animales, pero aún no han sido probados en estudios clínicos. Dichos fármacos incluyen: activadores del canal de calcio de la PC2 (triptolide), metformina, agonistas del receptor gamma activado por el proliferador peroxisómico, inhibidores o antagonistas de los transportadores y canales requeridos para la secreción de cloruro (inhibidores de CFTR), e inhibidores de la sintetasa de glucosilceramida.
Otros fármacos que han sido efectivos en ensayos preclínicos y que muestran posible utilidad para el tratamiento de la PQR en humanos incluyen inhibidores de Erb-B1 (receptor de factor de crecimiento epidérmico) y Erb-B2, Src, MEK, y quinasas dependientes de ciclinas.
Enfermedad renal poliquística autosómica recesiva
El enfermedad poliquística autosómica recesiva (ARPKD) es una forma frecuentemente grave de PKD que afecta los riñones y las vías biliares. Se ha estimado una incidencia de 1:20.000 nacidos vivos.38 Todas las formas típicas de ARPKD resultan de mutaciones en el gen, PKHD1. Este gen está localizado en el cromosoma 6p21.1-p12.37,38
Diagnóstico
El diagnóstico puede ser realizado en el periodo intrauterino, neonatal o en los primeros meses de vida, por medio de una ecografía renal que evidencie un aumento difuso del volumen renal bilateral. El oligohidramnios es un hallazgo común, y debido al bajo gasto urinario fetal, puede desarrollar la secuencia de Potter (hipoplasia pulmonar, fascies típicas y anomalías en extremidades).39
Además del riñón, también se encuentra compromiso hepático con hepatomegalia, incremento de la ecogenicidad y dilatación de los ductos biliares intrahepáticos. Aunque la fibrosis hepática está histológicamente presente desde el nacimiento, los hallazgos clínicos, radiológicos y de laboratorio pueden estar ausentes al momento del diagnóstico.4
Como presentación inicial, el 45 % de los pacientes debutan con anomalías hepáticas, y por otra parte, debido a la hipoplasia pulmonar, hasta un 30 % de los neonatos afectados fallece a causa de insuficiencia respiratoria,39 antes de que se haya arribado al diagnóstico de ARPKD, con el consecuente subregistro.2
La patología revela un compromiso renal simétrico y bilateral. Histológicamente, los riñones muestran un patrón de dilataciones fusiformes (microquistes menores a 4 mm de diámetro) radiados desde la médula hacia la corteza.40 Estudios de microdisección y localización tubular han demostrado que la enfermedad está confinada a los túbulos colectores en todos los niños afectados; una fase quística transitoria ocurre en los túbulos proximales fetales.41
Los casos menos graves presentan riñones palpables bilaterales, hipertensión arterial, hipostenuria, acidosis metabólica y falla renal progresiva. En cuanto al deterioro hepático, este puede resultar asintomático o progresara hipertensión portal.37
Los casos menos graves presentan riñones palpables bilaterales, hipertensión arterial, hipostenuria, acidosis metabólica y falla renal progresiva. En cuanto al deterioro hepático, este puede resultar asintomático o progresar a hipertensión portal.38
Tratamiento
Los neonatos con cuadros graves, requieren medidas de soporte vital por lo que es indispensable determinar el grado de afectación pulmonar y la evaluación de la función respiratoria, saturación de oxígeno, radiografía de tórax y exámenes paraclínicos pertinentes según el caso. Adicionalmente, se debe realizar ecografía renal, medición de la presión arterial, transaminasas, bilirrubinas séricas, albumina sérica, tiempos de coagulación y hemograma. Si se presenta oliguria o anuria, se debe iniciar diálisis peritoneal en los primeros días de vida. Si los riñones presentan un tamaño aumentado, algunos autores recomiendan nefrectomía unilateral o bilateral según el compromiso del paciente y la afectación de los órganos adyacentes involucrados.40
El manejo de la hipertensión arterial asociada se realiza con IECAs o ARA II. Las infecciones urinarias recurrentes son una complicación frecuente en este tipo de pacientes, por lo que se debe vigilar estrechamente su aparición, así como la presencia de nefrolitiasis asociada. Se debe evitar la administración de medicamentos simpaticomiméticos, agentes nefrotóxicos, AINEs, aminoglucósidos, cafeína, teofilina y bloqueadores de canales de calcio. En cuanto a la sobrevida de los pacientes, la tasa anual es del 71-75% a los 10 años y del 66% a los15 años. 41
Asesoría genética
Como ya se ha mencionado, esta patología se transmite por un mecanismo de herencia autosómico recesivo. En principio, los padres de un menor afectado son portadores obligados; tienen un riesgo del 25 % en cada embarazo de transmitir los alelos afectados y que nazca un afectado nuevamente.
Padres de un niño afectado
Los padres de un niño afectado son heterocigotos obligados (cada uno de ellos porta un alelo mutante). La probabilidad de tener otro hijo afectado es del 25 % en cada embarazo; la probabilidad de tener un hijo portador de un alelo mutado es del 50 % y la probabilidad que no porte ningún alelo mutado es del 25 %.
Los heterocigotos son asintomáticos, aunque es importante realizar ecografía renal en padres de niños con sospecha de ARPDK, para excluir la posibilidad de ADPKD. No existen datos sistemáticos disponibles de la sensibilidad y especificidad de la ecografía prenatal en el diagnóstico de ARPKD. Desde el punto de vista radiológico, los riñones quísticos y brillantes, cuando se detectan incidentalmente en una ecografía prenatal de rutina, son un dilema diagnóstico, ya que pueden ser producidos por diversas etiologías, con numerosas implicaciones para el pronóstico fetal y futuros embarazos.42
Si no existe historia familiar de ARPKD, pero en la ecografía prenatal se evidencian riñones quísticos y aumentados de tamaño, se recomienda realizar ecografía fetal de detalle y cariotipo para evaluar la presencia de anomalías cromosómicas u otras anomalías congénitas en el feto.42
La prueba molecular se puede realizar con el análisis de ADN de células fetales, por amniocentesis (entre la semana 15 y 18 de gestación) o a la semana 12 de gestación por biopsia de vellosidad coriónica.42 El diagnóstico genético pre implantacional está disponible para familias en las que se ha identificado la mutación causal de la enfermedad.41
Hijos de un afectado con ARPKD
Los hijos de un individuo afectado con ARPKD son todos heterocigotos obligados (portadores) de una mutación causante de la enfermedad. La frecuencia de portadores en la población general es de 1:70; por lo tanto, el riesgo que uno de sus hijos presente la enfermedad, depende de si su pareja es portador(a) de la mutación. Teniendo en cuenta la frecuencia de portadores en la población general, el riesgo sería del 0,7 %.43
Detección de portadores
Se realiza una vez se detecte la mutación en el probando. Si aún no se conoce la mutación enla familia, se realiza un análisis de ligamiento. Se debe hacer hincapié en que el momento ideal para determinar el riesgo genético, determinación del estatus de portador y la realización del test prenatal debe ser antes del embarazo, 28 en cuanto al pronóstico de los pacientes.44
Conclusiones
La Enfermedad Renal Poliquística es la enfermedad renal de origen genético más importante, y tiene dos patrones de transmisión hereditaria: la autosómica dominante y la autosómica recesiva, siendo la más grave la forma autosómica recesiva. Constituye una de las causas más significativas de ERC grado 5 bajo terapia sustitutiva renal.
Existe gran variabilidad en la expresión fenotípica de esta enfermedad, la cual no sólo depende de los loci afectados sino de otros factores desconocidos. La terapéutica actual se encamina hacia el control de los síntomas y en evitar la pérdida del parénquima renal mediante el control de las enfermedades y factores de riesgos asociados, fundamentalmente la hipertensión arterial.
La prevención de la enfermedad está basada en la asesoría genética. Actualmente existen numerosos ensayos clínicos prometedores, con fármacos que persiguen detener el desarrollo de los quistes en el riñón y de este modo conseguir la conservación del parénquima.
Referencias bibliográficas
1. Adeola T, Adeleye O, Potts JL et al. Thoracic aortic dissection in a patient with autosomal dominant polycystic kidney disease. J Natl Med Assoc.2001; 93:282-7.
2. Durán Álvarez Sandalio. Enfermedad renal poliquística autosómica dominante. Rev Cubana Pediatr [revista en la Internet]. 2007 Sep [citado 2014 Abr 14]; 79(3). Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75312007000300010&lng=es .
3. Guatibonza Y; et al. Actualidad de la Enfermedad Renal Poliquística. Univ. Méd. Bogotá (Colombia). 2013; 54 (1): 53-68.
4. Sociedad Ecuatoriana de Nefrología. Reporte 2013. Reporte 2013. Recolección de datos. [citado 2014 feb 12] http://sociedadecuatorianadenefrologia.org/wp-content/uploads/2014/05/editorial- revsen.pdf
6. Stengel B, Billon S, van Dijk PC. Trends in the incidence of renal replacement therapy for end stage renal disease in Europe, 1990-1999. Nephrol Dial Transplant. 2003; 18:1824-33.
7. Wilson P. Polycystic kidney disease. N Engl J Med. 2004;350:151-64.
8. Durán S. et al. Enfermedad Renal Poliquística Autosómica Dominante. Revista Cubana de Pediatría. 2007; 79 (3)
9. Coto E, Aguado S, Álvarez J, Menéndez Díaz MJ, López Larrea C. Genetic and clinical studies in autosomal dominant polycystic kidney disease type 1. J Med Genet. 1992; 29: 243-246.
10. Stengel B, Billon S, van Dijk PC. Trends in the incidence of renal replacement therapy for end stage renal disease in Europe, 1990-1999. Nephrol Dial Transplant. 2003; 18:1824-33.
11. Wilson P. Polycystic kidney disease. N Engl J Med. 2004; 350:151-64.
12. Schrier RW. Optimal careof autosomal dominant polycystic kidney dis- ease patients. Nephrology Carlton. 2006; 11:124-30.
13. Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA. 2006; 103:5466-71.
14. Klahr S, Breyer JA, Beck GJ, e tal. Di- etary protein restriction, blood pressure control, and the progression of polycystic kidney disease. J Am Soc Nephrol. 1995; 5:2037-47.
15. US Renal Data System. US RDS 2010. Annual Data Report: Atlas of chronic kidney disease and end stage renal disease in the United States [internet]. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2010.
16. Rodríguez R. Retardo en la progresión del daño renal en pacientes con insufciencia renal crónica estado 4: impacto de un programa de prevención en prediálisis. Asocolnef. 2007;1:10-21.
17. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guide- line for t e evaluation and management of chronic kidney disease. Kidney Inter. Suppl. 2013;3:1-150.
18. Park EY WY. Polycystic kidney disease and therapeutic approaches. BMB Re- ports. 2011 Jun;44:359-68.
19. Grantham JJ, Torres VE, Chapman AB, et al. Volume progression in poly cystic kidney disease. N Engl J Med. 2006;354:2122-30.
20. Harris PC, Rossetti S. Molecular diagnostics for autosomal dominant poly- cystic kidney disease. Nat Rev Nephrol. 2010;6:197-206.
21. Harris PC, Rossetti S. Determinants of renal disease ariability in ADPKD. Adv Chronic Kidney Dis. 2010;17:131-9.
22. Handa SP. Cardiovascular manifestations of autosomal dominant polycystic kidney disease in young adults. Clin Invest Med. 2006;29:339-46.
23. Seeman DJ. Ambulatory blood pressure correlates with renal volume and number of renal cysts in children with autosomal dominant polycystic kidney disease. Blood Press Monit. 2000;8:107-10.
24. Grantham JJ. Autosomal dominant polycystic kidney disease. N Engl J Med. 2008;359:1477-85.
25. Hateboer N, DijkMAv, Bogdanova N, et al. Comparison of phenotypes of polycystic kidney disease types1and 2.Lancet. 1999;353:103-7.
26. Lieske JC, Toback FG. Autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1993;3:1442-49.
27. Serra A L, Poster D, Kistler A D, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363: 820-9.
28. Torres V E, Harris P C, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287-301.
29. Torres V E. Treatment strategies and clinical trial design in ADPKD. Adv Chronic Kidney Dis. 2010;17:190-204.
30. Ravine D, Gibson RN,Walker RG, et al. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet. 1994;343:824-7.
31. Nicolau C, Torra R, Badenas C, et al. Autosomal dominant polycystic kidney disease types 1 and 2: assessment of US sensitivity for diagnosis. Radiology. 1999;213:273-6.
32. Ecder TSR. Hypertension in autosom dominant polycystic kidney disease: early occurrence and unique aspects. J Am Soc Nephrol. 2001;12:194-200.
33. Sarnak M.The effect of alower target blood pressure on the progression of kidney disease: long-term follow-up of the modification of diet in renal disease study. Ann Intern Med.2005;142:342-51.
34. Alam A, Perrone RD. Management of ESRD in patients with autosomal domi- nant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17:164-72.
35. International Advisory Board Meeting 2006:Abstracts. Nutritional therapy in pa- tients with chronic kidney disease: protein- restricted diets supplemented with keto/ aminoacids. Am J Nephrol.2006;26(Sup- pl.1):5-27.
36. Ong AC, Devuyst O. Towards the integration of genetic knowledge into clinical practice. Nephron Clin Pract.2011;118:c3-8.
37. Pagon RA, Bird TD, Dolan CR,et al., editors. Genere views. Seattle (WA): University of Washington; 1993.
38. Pei Y. Practical genetics for autosomal dominant polycystic kidney disease. Nephron Clin Pract. 2011;118:c19-30.
39. Grantham J, MulamallaS, Swenson Fields K. Why kidneys fail in autosomal dominant polycystic kidney disease. Nat Rev Nephrol. 2011;7:556-66. DOI:10.1038/nrneph.2011.109.
40. Beevers C. Curcumin inhibits the mammalian target of rapamycin-mediated signaling pathways in cancer cells. Int J cancer. 2006;119:757-64.
41. Alam A, Perrone RD. Management of ESRD in patients with autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17:164-72.
42. WalzG, BuddeK, Mannaa M, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:830-40.
43. Rossetti S, Consugar M B, Chapman A B, et al. CRISP Consortium. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007;18:2143-60.
44. Zerres K, Mucher G, Becker J, et al. Prenatal diagnosis of autosomal recessive polycystic kidney disease (ARPKD): molecular genetics, clinical experience, and fetal morphology. Am J Med Genet. 1998;76:137-44.
45. Torres VE. Vasopressin antagonists in polycystic kidney disease. Kidney Int 2005;68:2405-18.
46. Wang X, Wu Y, Ward CJ, et al. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 2008;19:102-8.
47. Irazabal MV, Torres VE. Poliquistosis Renal Autosómica Dominante. 2011 Revista Nefrología. Órgano Oficial de la Sociedad Española de Nefrología. Nefrologia Sup Ext 2011;2(1):38-51.
Recibido: julio de 2014.
Aprobado: diciembre de 2014.
1 Especialista de I grado en Medicina Interna. Médico Tratante de la Unidad Renal Dialibarra Cia. Ltda. Universidad Técnica del Norte. Ibarra. Ecuador.
2 Licenciada en Enfermería. MSc. en Docencia Educativa e Investigación Universitaria. Universidad Técnica del Norte. Ibarra. Ecuador.
3 Licenciada en Terapia Física. MSc. en Gerencia de la Salud. Universidad Técnica del Norte. Ibarra. Ecuador.
4 Licenciada en Enfermería. MSc. en Salud Familiar. Universidad Técnica del Norte. Ibarra. Ecuador.
Autor de correspondencia: Pedro Cena Rivero. Universidad Técnica del Norte. Ibarra. Ecuador.